


INTRODUCTION
Primary sarcoma of the kidney is a rare neoplasm that occurs at 1ŌĆō3% the rate of whole malignant renal cancer1,2). Synovial sarcoma is a soft tissue tumor that develops predominantly in the extremities near the para-articular, joint capsule, bursa, and tendon sheath in adolescents and young adults. The occurrence of the tumor in various extra-articular sites, such as the head and neck, chest, abdominal wall, and retroperitoneum, is extremely rare3). Primary renal synovial sarcoma is extremely rare and has poor prognosis. We report a case of synovial sarcoma of the kidney with metastasis to the lung, which was completely remitted by combination chemotherapy.
CASE REPORT
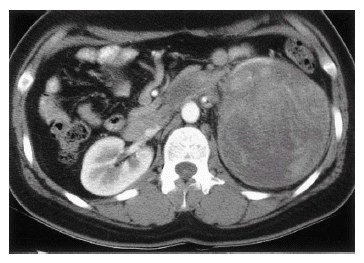
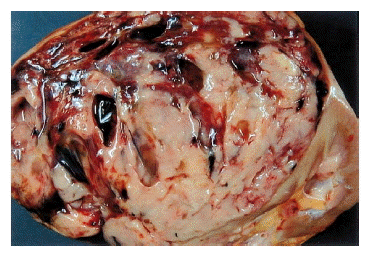
A 32 year-old woman was admitted due to intermittent abdominal pain which had persisted for one month. She had neither any remarkable past medical history nor family history. On physical examination, there was tenderness on the left upper quadrant of the abdomen; however, there was no palpable mass in the abdomen or no tenderness in both costovertebral angles. Laboratory data revealed that the lactate dehydrogenase (LDH) had risen to 1,114 IU/L. The others were within normal limits. Chest plain film showed streaky radiopaque densities on the right upper lobe, considered a scar of the inactive tuberculosis. Abdominal ultrasonogram showed a well-defined mixed echogenic mass on the left kidney. Abdominal computed tomography (CT) scan showed a well-defined mass with heterogenous enhancement on the lower pole of the left kidney with hilar infiltration, measured to be 12├Ś10 cm in size (Figure 1). Small-sized hilar lymph nodes and a thrombus in the left renal vein were observed. On day 23, a left radical nephrectomy was performed with left renal vein thrombectomy. A gross specimen was obtained from the left kidney of the patient, a 13├Ś12├Ś11 cm-sized mass on the lower pole of the kidney. The cut surface of the mass was grayish-white mixed with focal necrosis and hemorrhage (Figure 2).
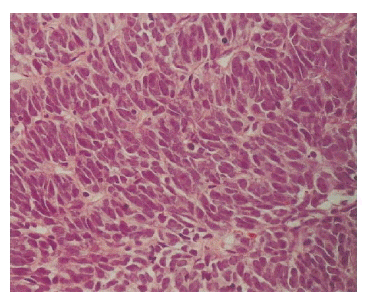
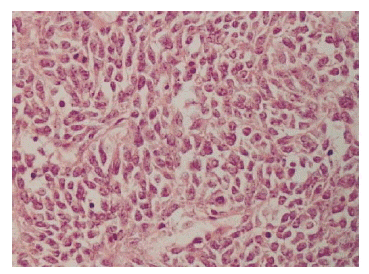
A light microscopic finding revealed a highly cellular tumor composed of mitotically active (to the degree about 30 mitoses/10 high power field), atypical plump spindle cells, and indistinct cell borders arranged in short and intersecting fascicles, mainly involving the renal pelvis with extension to the renal cortex and capsule (Figure 3). Malignant cells were not found in the dissected lymph nodes or in the renal vein.
On immunohistochemical stains, the tumor cells showed positive reaction to vimentin (Figure 4), but showed negative reactions to cytokeratin, epithelial membrane Antigen (EMA), S-100 protein, and CD34. There were no extrarenal manifestations, and we diagnosed the tumor as spindle cell-type monophasic synovial sarcoma, which had originated from the kidney.
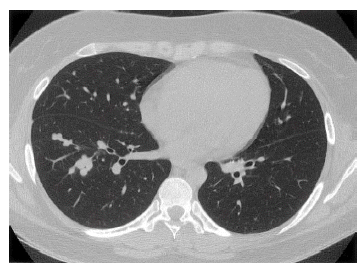
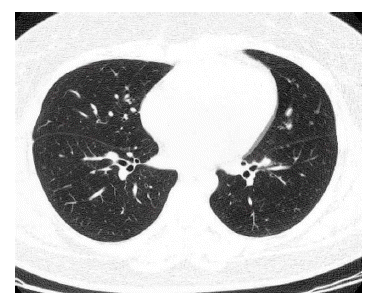
Four months later, chest radiography showed a number of small nodular opacities considered as hematogenous metastasis. Chest CT scan showed variable sized, nodular opacities with vascular connection in the entire lobe of the right lung (Figure 5). Percutaneous transthorasic needle biopsy (PTNB) on the right lower lung nodule was performed, and the pathologic finding was consistent with renal synovial sarcoma (Figure 6). She had received combination chemotherapy with 28-day cycles of ifosfamide 2,500 mg/m2 on day 1 through 3 plus doxorubicin 60 mg/m2 on day 1. After the second cycle of chemotherapy, follow-up chest CT scan showed smaller-sized metastatic nodules on the right lung field, and the state of the response was partial remission. Toxicity was scored according NCl criteria. During chemotherpay, grade 2 neutropenia and grade 1 thrombocytopenia was developed. Non-hematologic toxocities were grade 1 nausea and grade 3 alopecia. Planed doses were administered without dose reduction. After 6 cycles of chemotherapy, complete remission was achieved (Figure 7).
DISCUSSION
Sarcoma originating from the kidney is very rare. Sarcoma is classified according to histologic type: leiomyosarcoma, rhadomyosarcoma, liposarcoma, fibrosarcoma, malignant fibrous histiocytoma, hemagiopericytoma, and osteosarcoma2,4,5).
The most common renal sarcoma is leiomyosarcoma, which accounts for 40ŌĆō60% of the all reported cases, while primary synovial sarcoma in the kidney is extremely rare4ŌĆō6). Clinicopathologically, synovial sarcoma is a well-defined neoplasm, and the 4th most common source accounts for approximately 5ŌĆō10% of the all soft tissue sarcoma cases4). Synovial sarcoma occurs predominantly in the extremities near the para-articular, joint capsule, bursa, and tendon sheath, but has been reported in a variety of unusual locations in which there is no synovial structure, such as the head and neck area, ventricle of the brain, peripheral nerve, heart and pericardium, lung and pleura, mediastinum, and retroperitoneum3, 4, 7, 8). Renal synovial sarcoma was first reported by Faria et al. in 1999, and is a clinically and morphologically well-defined soft tissue tumor5). Many theories insist synovial sarcoma originates from either epithelial cells, nerve cells, or mesenchymal cells, but its histogenesis is still unknown2ŌĆō5). It occurs most often in adolescents and young adults like other soft tissue neoplasms1, 2). Histologically, it is subclassified into monophasic, bipahasic, and poorly differentiated types. The biphasic type is an admixture of epithelial cells and spindle cells and is easily recognized. On the other hand, the monophasic type is composed of either only epithelial cells or spindle cells, and it is difficult to distinguish from other sarcoma such as fibrosarcoma, hemangiopericytoma, or malignant peripheral nerve sheath tumor (MPNST, malignant schwannoma)3, 4, 9, 10).
Immunohistochemical finding is useful for diagnosing and distinguishing synovial sarcoma from other tumors. Hemangiopericytoma shows abundant vasculatures with no spindle cell and negativity for cytokeratin8). A MPNST resembles the monophasic fibrous type of synovial sarcoma, but, immunohistochemically, it shows a focal weak positive reaction to S-100 protein and a negative reaction to cytokeratin. A fibrosarcoma is composed of long spindle cells, and shows an arrangement of the fibroblasts in distinct intersecting fascicles, in what is called a herringbone pattern3,6,8). In this case, there is no extrarenal manifestation by clinical or radiologic finding. Additionally, fibrosarcoma showed positive reaction to vimentin, which could be seen in monophasic fibrous synovial sarcoma, and negative reactions to muscle-specific action, desmin, and S-100 protein, which is consistent with spindle cell-type monophasic fibrous synovial sarcoma. By light microscopic and immunohistochemical findings, this case was compatible with spindle cell-type monophasic synovial sarcoma of soft tissues and accordingly, we diagnosed this case as primary renal spindle cell-type monophasic synovial sarcoma. Mesothelioma was excluded due to the negativity for CD34. However, without molecular confirmation, differentiating between synovial sarcoma and congenital mesoblastic nephroma or clear cell sarcoma of the kidney is difficult due to their similarities in histological appearance. Although molecular analyses by RT-PCR to detect SYT-SSX fusion transcripts associated with the characteristic translocation (X; 18) of synovial sarcoma could confirm diagnosis, synovial sarcoma is not completely excluded by a negative RT-PCR1,6,11). SYT-SSX fusion proteins may trigger synovial sarcoma development, and chromosomal translocation (X;18)(p11.2;q11.2) is found in 90% of all reported cases6,9).
Taken together, despite some questions still unanswered, we think that the current case is consistent with synovial sarcoma.
The mainstay of treatment for all soft tissue sarcomas is surgical excision. Extensive resection is used because positive histologic margins at the moment of resection are poor prognostic factors2,12). Nonetheless, 30 to 50% of all patients who underwent adequate surgical resection are reported to have faced distant metastasis, usually to the lung or liver2). Radiation therapy was not settled until recently, even though the use of X-ray for the treatment of sarcoma was first proposed in 19022,13). However, it is clear that combination chemotherapy with doxorubicin and ifosfamide shows responses, and more intensive therapy may lead to higher rates of survival2). Although characterized by slow growth, synovial sarcoma sometimes results in poor long-term survival because of local recurrence and distant metastasis, especially to the lung13,14). In this case, distant metastasis to the lung developed four months after complete resection of the tumor. Interestingly, complete remission of the hematogenous metastasis was induced after completion of the sixth cycle of combination chemotherapy with doxorubicin and ifosfamide. However, hematogenous metastasis of the lung recurred after 5 months. After the sixth cycle of salvage chemotherapy with ifosfamide 2,000 mg/m2/day (total 4000 mg), etoposide 100 mg/m2/day (total 500 mg), and cisplatin 20 mg/m2 (total 100 mg), she was still alive with a stable disease state. Continuous follow-up is necessary.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





