


INTRODUCTION
Rapid, effective anti-platelet medication plays a crucial role in the prevention of stent thrombosis. Combined anti-platelet treatment with aspirin and clopidogrel significantly reduced stent thrombosis in patients undergoing coronary stenting. The CURE (Clopidogrel in Unstable angina to prevent Recurrent Events) study showed that combinations of clopidogrel and aspirin therapy reduced a composite of cardiovascular death, myocardial infarction, and stroke by 20% when compared to aspirin alone1). However, aspirin resistance2) and interindividual variability in the platelet inhibitory response to clopidogrel were noted in patients undergoing elective coronary stenting, especially in the context of high pretreatment platelet activity3). Clopidogrel resistance is associated with an increased risk of recurrent atheroembolic events in patients with acute myocardial infarction4). Moreover, P-selectin, which is expressed exclusively by platelet activation, upregulates tissue factor in monocytes, promotes fibrin deposition, and leads to the accumulation of leukocytes in areas of vascular injury associated with thrombosis and inflammation5, 6). The animal study illustrated that P-selectin deficiency reduces atherosclerotic lesion formation and neointimal formation after arterial injury7, 8). Therefore, an alternative pharmacological strategy is needed to produce more potent anti-platelet effects, especially in poor clopidogrel responders. Cilostazol is a potent anti-platelet agent which selectivelyinhibits phosphodiesterase III9). Previous studies showed that cilostazol had similar anti-platelet effects with fewer serious adverse effects than ticlopidine in preventing stent thrombosis. It was also shown that cilostazol reduced restenosis after coronary balloon angioplasty and coronary stenting10, 11). However, whether cilostazol has synergistic anti-platelet effects when combined with clopidogrel and aspirin remains unknown. We studied the change in platelet activation after 2 weeks of cilostazol therapy in patients already taking clopidogrel and aspirin due to coronary stent by measuring markers of platelet activation (P-selectin and activated GPIIb/IIIa) using whole blood flow cytometry. We also studied the influence of CRP on platelet activation.
MATERIALS AND METHODS
Subjects
From March 2004 to May 2004, 47 patients who had been taking clopidogrel with aspirin for more than one month subsequent to coronary stent due to ischemic heart disease were included to this study. The criteria for exclusion were contraindication to anti-platelet agents, cancer, chronic inflammatory disease, known bleeding tendencies, inability to follow the study protocol, thrombocytopenia (<100,000/m3), severe hepatic or renal dysfunction (serum creatinine >2 mg/dL), and use of GPIIb/IIIa inhibitors. Table 1 gives the demographic and clinical data of the study group. Cilostazol 50 mg/BID was administered to all patients. Statin and other drugs were also given if indicated.
Blood sampling and flow cytometric analysis
Blood samples were taken after 12 hrs of fasting prior to cilostazol administration (baseline) and 14 days after beginning cilostazol treatment for hsCRP (mg/dL) and markers of platelet activation. We used a 2-syringe technique to reject the first few of the sample to minimize artifactual platelet activation. Blood was collected in evacuated container tubes containing 1:1 3.8% trisodium citrate and HEPES buffer (HEPES 10 mM, NaCl 150 mM, KCL 5 mM, MgSO4 1 mM, glucose 10 mM, pH 7.4, 290 mosM) to minimize platelet aggregation It was then inverted 3 to 5 times for gentle mixing.
Briefly, in order to evaluate P-selectin expression, 10 uL of blood-HEPES mixture was diluted in 50 uL of HEFES buffer and each fluorescein isoethiocyanate (FITC)-labeled monoclonal antibody against P-selectin (CD62P.FITC, Becton-Dickinson, NJ, USA) and monoclonal antibody against GP1b were added to identify platelets (CD42b, PE, Beckton-Dickinson). After careful mixing and incubation in the dark for 15 min at room temperature, 350 uL of 1% formaldehyde (HEFES 99 : Formalin 1) was added for fixation. This fixation process was shown to preserve P-selectin (CD62P) and GPIIb/IIIa activity for at least 24 hours at room temperature. The fraction of circulating platelets expressing the activation markers p-selectin and GPIIb/IIIa was determined by flow cytometric analysis as previously described12). Samples were then evaluated in the flow cytometer (Colter Epics XL, USA). In order to evaluate GPIIb/IIIa, 10 Ul of blood-HEFES mixture was diluted in 50ul of HEPES buffer. Saturating concentrations (20ul) of the antibody that recognizes GPIIb/IIIa (PAC-1.FTIC) and CD42b (PE) was added. The PAC-1 fluorescein isothiocyanate (FTIC) antibody binds completely at the activated GPIIb/IIIa receptor complex. The samples were then analyzed in the flow cytometer. For standardization and alignment, the flow cytometer was calibrated daily with Flowcheck microbeads (Beckman coulter, USA). Acquisition data analysis was performed using the EXPO32 ABC analysis program. Results are expressed as a percentage of 5000 platelets.
Data analysis
Data are shown as mean standard deviation. We used a paired sample t-test and correlation analysis to compare the change in P-selectin expression and activated GPIIb/IIIa on the platelet surface before and after cilostazol treatment in the total patients. Baseline characteristics were analyzed by chi-square test between the P-selectin decreased and non-decreased groups after cilostazol treatment. To assess the effect of pretreatment platelet activity on response to cilostazol, patients were divided into high, moderate, low groups according to baseline P-selectin expression. Comparisons were made between groups by 1-way ANOVA. We used the software package SPSS for windows, version 11.5.0 for Windows (SPSS Inc); p-values less than 0.05 were considered significant.
RESULTS
Patient Data
Completed platelet studies were performed at baseline and after cilostazol treatment for forty-seven patients. The demographics of these 47 patients are shown with respect to the response of P-selectin change in the Table 1.
Prevalence of cardiovascular risk factors and statin administration did not differ significantly between groups. Follow up at 30 days revealed no cases of Q wave myocardial infarction, stent thrombosis, target vessel revascularization, cerebrovascular ischemic events, or death.
CD62p (P-selectin) expression after cilostazol treatment
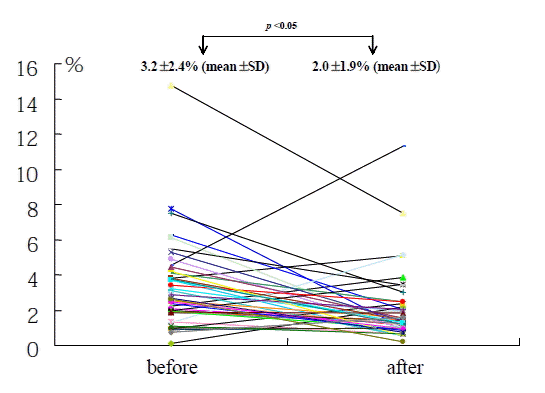
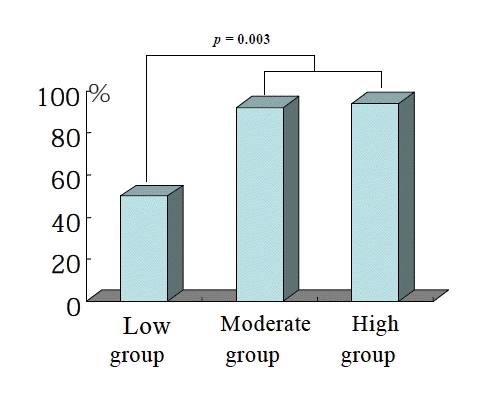
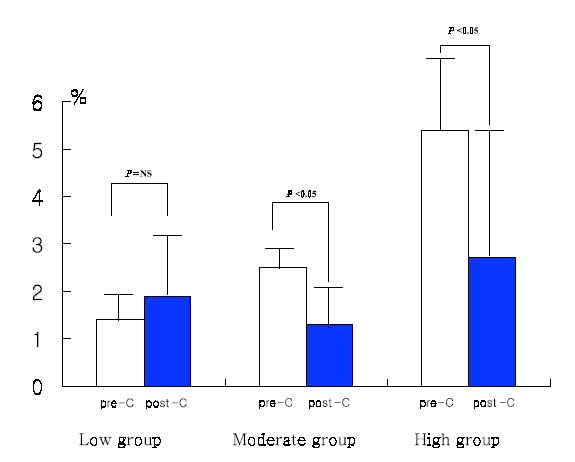
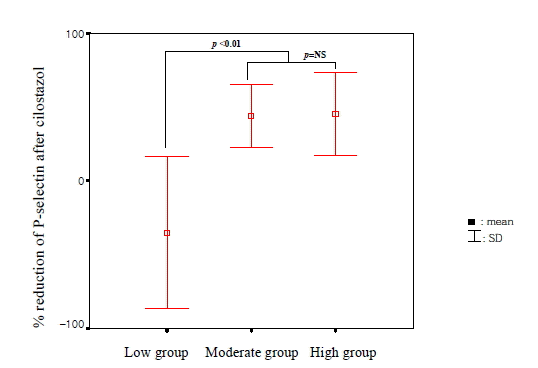
Baseline P-selectin expression was 3.2±2.4% and fell to 2.0±1.9% after 14 days of cilostazol treatment in the total patient population (p=0.04) (Figure 1). Among 47 patients, 36 (76.6%) showed decreased P-selectin expression levels after cilostazol treatment as compared to baseline. There was no significant difference in age, sex, smoking, DM, hypercholesterolemia, and hsCRP between the P-selectin increased group and the non-increased group (Table 1). Baseline P-selectin expression levels were correlated to post-cilostazol treatment P-selectin expression (r=0.36, p=0.013). When subjects were divided into tertiles based on levels of P-selcetin level positivity before cilostazol treatment (low group, mean 1.42, 0 to 1.96%, n=16; moderate group, mean 2.53, 1.97 to 3.43%, n=14; high group, mean 5.48, 3.44 to 14.77%, n=17), P-selectin expression levels decreased after cilostazol treatment in more patients of the moderate and high groups than those of low group (low group : 50%, middle group : 92%, high group : 94%, p=0.003) (Figure 2). The amount (low group; 1.4±0.5 to 1.9±1.3%, p=NS, moderate group; 2.5±0.3 to 1.3±0.3%, p<0.05, high group; 5.42.7 to 2.7±2.8%, p<0.05) (Figure 3) and % reduction of P-selectin expression levels after cilostazol treatment (low group; −35.19±9.2%, moderate group; 44.0±39.5%, high group; 45.2±58.1%, p<0.05) (Figure 4) were greater in the moderate and high groups than that of the low group.
PAC-1 (activated GPIIb/IIIa) expression after cilostazol treatment
Baseline PAC-1 expression was not increased after cilostazol treatment (from 13.5% to 17.6%, p=NS). No difference of age, sex, serum cholesterol and hsCRP level was observed between the PAC-1 increased group (n=27, 57%) and the non-increased group (n=27, 43%)
Relation between hsCRP and platelet receptor activation
hsCRP was unchanged after cilostazol treatment (baseline; 0.30±0.38 mg/dL, post-cilostazol treatment; 0.38±0.69 mg/dL, p=NS). Baseline levels of hsCRP correlated with post-cilostazol treatment hsCRP levels (r=0.76, p<0.001). No significant correlation existed between platelet activation markers (P-selectin and activated GPIIb/IIIa) and hsCRP at either baseline or post-cilostazol treatment.
DISCUSSION
The main hypothesis of the present investigation was that in patients with coronary stenting, cilostazol treatment, in addition to clopidogrel and aspirin, might further reduce platelet activation below the level achieved by aspirin and clodpidogrel alone. This study revealed that an additional decrease in platelet P-selectin expression was induced by short term treatment of low dose cilostazol (100 mg/BID) to those who had already been given clopidogrel and aspirin. The present study is the first to demonstrate that triple therapy (cilostazol+clopidogrel+aspirin, C+A+C) is more effective in suppressing platelet activation than conventional therapy (clopidogrel+aspirin, C+A). This effect is noted mainly in patients with relatively high platelet activity despite more than 1 month of aspirin and clopidogrel administration.
Aspirin and Clopidogrel resistance
The presence of aspirin treatment failures in several disease states led to the concern that selected patients may be resistant to the effects of aspirin. Prevalence of aspirin resistance varied by disease condition and platelet function methodology12). Evidence exists regarding a link between aspirin resistance and clinical events such as stroke13, 14), and cardiovascular mortality15). Patients undergoing non-urgent PCI are at increased risk of myonecrosis when they are determined to be aspirin-resistant using a point-of-care assay, compared with those who are aspirin-sensitive16). More recently, variable platelet and potential resistance to thienopyridines has emerged. Studies have shown poor clinical outcomes in patients with clopidogrel resistance3, 17). Clodpidogrel reduces platelet activation via ADP receptor-dependent pathways. Previous experimental studies in both animals and humans, as well as large scale clinical trials, have demonstrated that a combination of aspirin and clopidogrel results in a combined inhibition of cyclooxygenase and ADP effects, and provides marked enhancement of antethrombotic efficacy with atherosclerotic vascular disorders1, 18–20). However, interindividual variability in the platelet inhibitory response due to clopidogrel occurs in patients undergoing elective coronary stenting, especially in high pretreatment platelet activity3). Interindividual variability in response to clopidogrel may affect clinical outcomes4, 21). Moreover, patients with symptomatic coronary artery disease who are undergoing coronary stenting are frequently given clopidogrel in conjunction with the HMG-CoA reductase inhibitor atorvastatin. This may decrease anti-platelet activity of clopidogrel despite adequate treatment with clopidogrel20), although reproduction of the long term clinical impact of these observations in larger datasets was necessitated. These results suggest that therapies must be found which can overcome the resistance to platelet aggregation inhibition. Most previous studies empirically defined clopidogrel resistance by measuring the amount of reduction in aggregation in response to ADP, although easily performed, reproducible ways to measure platelet aggregation should be found. In the present study, we assessed the expression of an established marker of platelet activation, by the P-selectin specific antibody (anti-CD62P) and the activated GPIIb/IIIa specific antibody (PAC-1), via whole blood flow cytometric analysis as an alternative to platelet aggregation. Flow cytometric analysis of platelets is a relatively new, powerful technique which is developing into a useful clinical tool. Whole blood flow cytometry has the advantage of directly analyzing individual platelets with a high degree of sensitivity and minimal artificial platelet activation23). We also empirically divided patients into low, moderate, or high P-selectin expression groups, according to the degree of pretreatment P-selectin expression.
P-selectin expression and cilostazol
In our study, cilostazol enhanced the suppression of P-selectin in moderate and high pretreatment P-selectin expression groups, while it did not suppress GPIIb/IIIa activation. These results suggest that cilostazol more strongly suppressed the expression of P-selectin on the platelet membrane surface by α-granule release than the conformational change of GPIIb/IIIa. P-selectin is a component of the α-granule membrane of resting platelets which is only expressed on the platelet surface membrane after α-granule secretion24). Cilostazol is a 2-oxo-quinoline derivative with antithrombotic, vosodilator, antimitogenic, and cardiotrophic properties. The compound is a potent inhibitor of phophodiesterase (PDE) 3A, the isoform of PDE3 in the cardiovascular system. PDE inhibition by cilostazol increases cAMP level, leading to the activation of protein kinase C, which induces a decrease in intracellular Ca++, resulted in suppression of platelet -granule release25). This inhibition of α-granule release by cilostazol might diminish P-selectin expression more than GPIIb/IIIa, as is shown in the present study.
Importance of P-selectin inhibition
P-selectin initiates the capture and rolling of circulating leukocytes at sites of inflammation and atherosclerosis26). The formation of leukocyte-platelet conjugates is mainly mediated by the binding of P-selectin glycoprotein ligand-1 (PSGL-1)27). The association of platelet-leukocyte interactions with acute coronary syndrome (ACS) has been well-demonstrated in various clinical settings28–32). The high proportion of platelet-monocyte conjugates in ACS suggests that they may play an important role in the mechanism of ACS, although a cause-effect relationship remains to be documented and clopidogrel attenuates the formation of platelet-monocyte and platelet-neutrophil conjugates in ACS33). Animal study illustrated that deficiencies in P-selectin reduce athersclerotic lesion formation and neointimal formation after arterial injury. Coronary stenting induced a release of adhesion molecules. P-selcetin and P-selectin mediated platelet-leukocyte interaction has a crucial role in the development of stent restenosis33, 34). In the Cilostazol for restenosis trial (CREST)35), cilostazol treatment with clodpidogrel and aspirin resulted in a significantly lower binary restenosis rate, as compared to placebo-treated patients. Actual P-selectin change of triple therapy (C+A+C) compared to conventional antiplatelet therapy (C+A), however, was not illustrated in humans before the present study. These results suggests that lower restenosis rates with anti-platelet triple therapy in the CREST study might be influenced by more effective inhibition of platelet P-selectin expression.
Study limitations and conclusion
The main objective of our study was to evaluate the synergistic effects of cilostazol with aspirin-clopidogrel on platelet activation. Unfortunately, the present study was a non-randomized, non-controlled study. Therefore, several factors which could influence platelet activation, such as natural change of platelet function as time goes by, artificial decrease of platelet activation by statin, inflammation, or acute plaque rupture, cannot be excluded. However, study subjects remained in a stable condition for more than one month after coronary stenting and a platelet activation test was completed at only 2 weeks of cilostazol administration without any remarkable clinical events observed. Moreover, it was known that steady state platelet activation was obtained after 5 days of clopidogrel administration, and lasted for more than one month in a previous study36). We believed that these aspects could minimize unexpected change to platelet activation. The observation period in our study is too short and the number of patients too small to investigate clinical effectiveness. Investigation of clinical effectiveness, however, was not our purpose. We were interested in platelet function with triple anti-platelet therapy and suggested that triple anti-platelet therapy would be an effective anti-platelet regimen, especially in patients responding poorly to clopidogrel. Therefore, this study does not allow inference regarding the clinical or prognostic value of triple anti-platelet therapy (cilostazol+clopidogrel+aspirin). This necessitates a large clinical trial. In conclusion, additional cilostazol treatment to conventional anti-platelet therapy (clopidogrel+aspirin) after coronary stenting provides more effective suppression of platelet P-selectin expression in patients with relatively high pre-treatment platelet activity, suggesting that cilostazol may prevent adverse clinical outcomes in poor clopidogrel responders.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





