


INTRODUCTION
Most identified lymphomatous infiltrations of the liver are a result of secondary involvement of widespread non-Hodgkin's lymphoma. According to the diagnostic criteria for a primary hepatic lymphoma (PHL), suggested by Caccamo et al.1) the lymphoma is confined to the liver with no evidence of lymphomatous involvement of the spleen, lymph nodes, bone marrow, or other lymphatic organs. PHL is very rare and there is no consensus on the best approach for management2). In Korea, 14 cases of PHL, have been reported since 19793-9). The most common diagnosis for a PHL is diffuse large B-cell lymphoma (DLBL)10, 11). In addition, there have been a few reports of primary hepatic mucosa-associated lymphoid tissue (MALT) lymphomas. Here we report a case of primary hepatic extranodal marginal zone B-cell lymphoma of MALT which was successfully treated with radiotherapy alone.
CASE REPORT
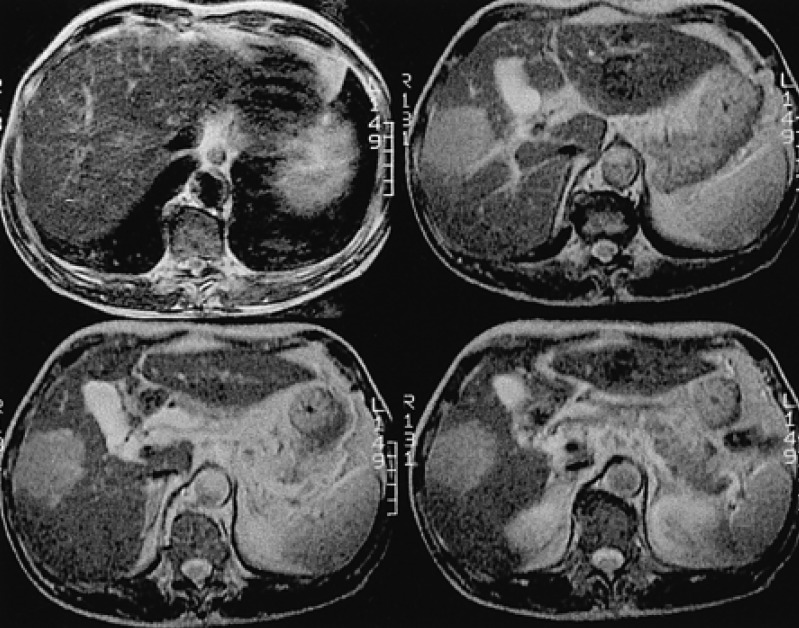
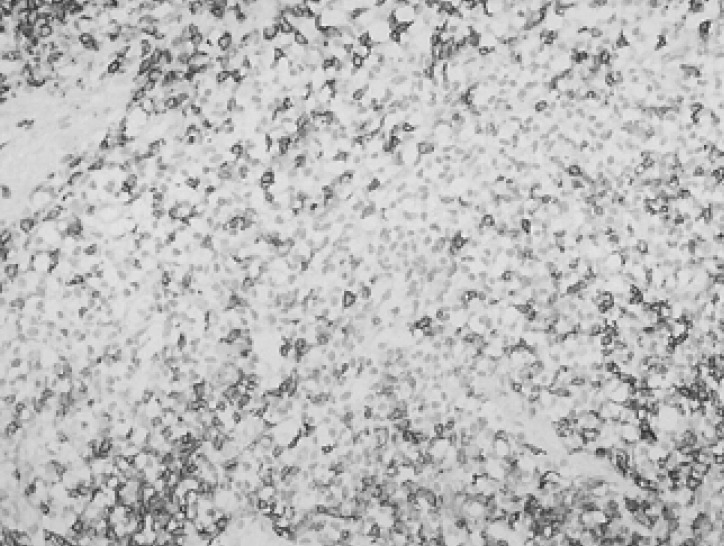
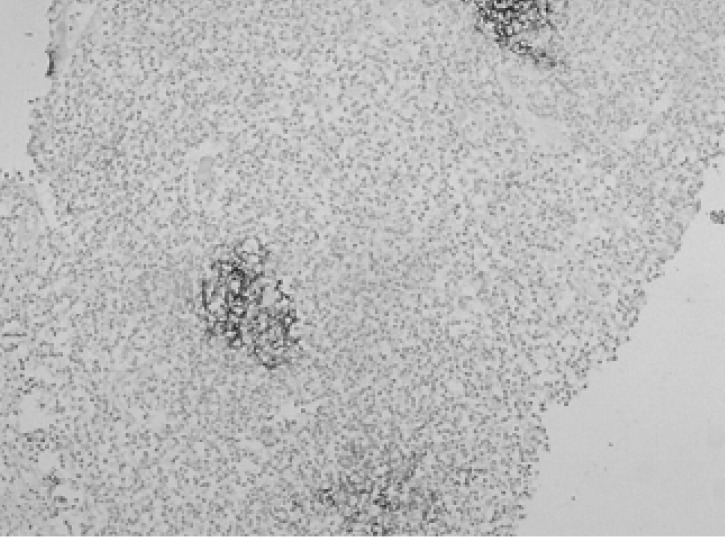
A 67-year-old man, who was undergoing treatment for a bleeding duodenal ulcer, was admitted to our hospital for evaluation of a liver mass incidentally found on abdominal ultrasonography. The patient had a past medical history of angina pectoris, drug induced hepatitis, myocardial infarction, congestive heart failure and old pulmonary tuberculosis. The family history was none contributory. The patient complained of general weakness, easy fatigability and anorexia. He had no complaints of abdominal pain, weight loss, fever or night sweats. The blood pressure was 125/90 mmHg, pulse rate 90/min and body temperature 36Ōäā. He appeared chronically ill and had an alert mental status. There was no tenderness on abdominal examination. Hepatomegaly of two fingers breadth was noted below the right costal margin; there was no ascites. His spleen and lymph nodes were not enlarged. Laboratory blood tests showed a hemoglobin of 12.8 g/dL, hematocrit 37%, white blood cell 5,400/┬ĄL with 42.8% neutrophils, platelet count 122,000/┬ĄL, blood urea nitrogen 14.2 mg/dL, creatinine 1.4 mg/dL, total protein 7.8 g/dL, albumin 3.8 g/dL, total bilirubin 0.8 mg/dL, alkaline phosphatase 53 IU/L, alanine aminotransferase (ALT) 30 IU/L, aspartate aminotransferase (AST) 35 IU/L, gamma glutamyl transferase (r-GT) 53 IU/L, lactate dehydrogenase (LDH) 250 IU/L (within normal range), prothrombin time 23 sec (98% of normal) and alpha-fetoprotein 3.5 ng/mL (within normal range). Hepatitis B surface antigen (HBsAg), antibody against hepatitis B surface antigen (anti-HBs) and antibody against hepatitis C (anti-HCV) were all negative. Abdominal magnetic resonance imaging (MRI) showed a well defined homogeneous mass 5.7├Ś4.8 cm in size at segment 5 (Figure 1). The following neck and chest computerized tomography (CT) scan showed no sign of other organ involvement or lymphadenopathy. Abdomen-pelvic CT scan showed no evidence of splenomegaly or lymphadenopathy. The whole body bone scan (WBBS) had no evidence of bone involvement. Gallium scan also showed no abnormal radioactivity. There were no malignant infiltrations noted on the bone marrow biopsy. Additional diagnostic evaluation included hepatic angiography and ultrasonograpy guided liver needle aspiration biopsy, using the gun shot approach, of the hypoechoic lesion in the right liver lobe. Hepatic angiography did not show any stained tumor, and lipiodol injection failed. Liver biopsy histology showed a severely distorted architecture due to lymphoreticular cell infiltration of the portal tract (Figure 2). The cells had the following features: they were uniform in size, had a slightly irregular outline and showed an increased nuclear-cytoplasmic ratio. They showed no particular arrangement representative of a specific organ. The cells expressed CD20 (Figure 3) with a low Ki-67 labeling index supporting the diagnosis of a low grade tumor (Figure 4). The cells had no immunoreactivity for CD5 and CD23 (Figure 5, 6). The final pathologic diagnosis was an extranodal marginal zone B-cell lymphoma of MALT according to the WHO classification. The patient underwent radiotherapy with a total dose of 4,140 cGy (1,980 cGy on the entire liver and an additional 2,160 cGy focused on the tumor area).
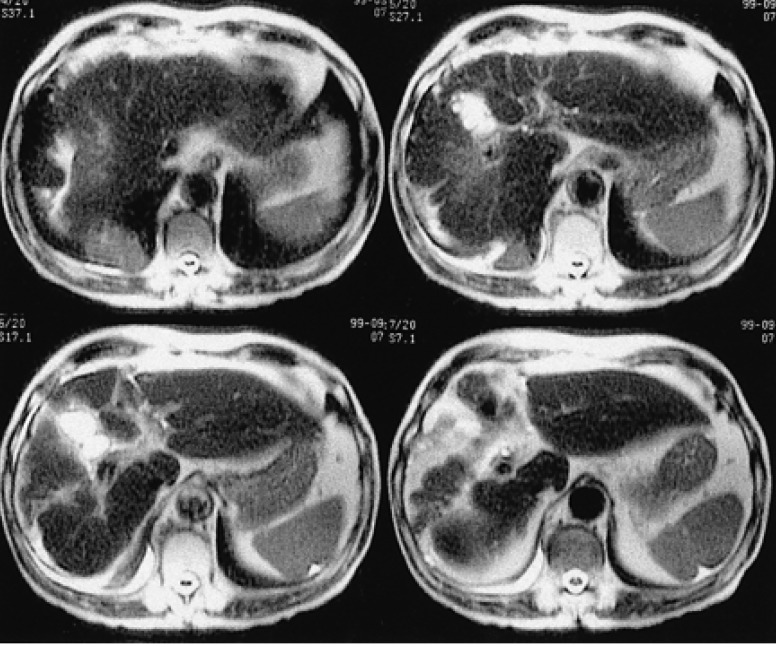
Abdominal ultrasonography, performed 5 years after radiotherapy, revealed complete remission of the lymphoma in the liver. In addition, the liver MRI, studied 5 years after radiotherapy, showed fibrous contraction due to post radiation change of right lobe of the liver. There was no evidence of tumor recurrence in the liver parenchyma (Figure 7). However, a newly developed well-enhancing solid mass, highly suggestive of a malignancy, at the posterior basal segment of right lower lobe of lung, was incidentally discovered by MRI at this time.
Six years after diagnosis, he was readmitted due to complaints of dyspnea. The chest X-ray, chest CT scan and Whole body bone scan showed progression of the mass from the right lower lobe of the lung, pleural effusion, and multiple bone metastases. The patient refused additional diagnosis and treatment, and died as result of an undefined disease of the lung.
DISCUSSION
Primary hepatic lymphoma (PHL) is defined as a lymphoma localized and limited to the liver1). This type of tumor is very rare representing less than 1% of all extranodal non-Hodgkin's lymphoma10, 11). In a review of 90 patients with PHL, conducted by Lei10) PHL was noted to occur primarily in middle-aged men (M:F=2.3:1); the most frequent presenting complaint was abdominal pain. The symptoms of fever and weight loss occur in about one-third of the patients. Night sweats are less common and occur in about 10% of patients2, 10, 11). Objective findings on examination include hepatomegaly in at least 50% of patients, with icterus noted in only about 10~20%2). The etiology of PHL is unknown; however, several possible etiologic factors have been proposed. HCV or HBV infection is found in 20~60% of patients with PHL2). The frequent association with HCV or HBV suggest that this virus may play a role in the pathogenesis of PHL. Our patient had no HCV or HBV infections. PHL may present radiologically as a solitary liver mass or as multiple lesions within the liver. The most common presentation is a solitary lesion, which occurs in about 55~60% of cases, followed by multiple lesions in about 35~40% of patients. Diffuse hepatic infiltration is rare and has a worse prognosis2). The predominant histology of the PHL is DLBL (46%); a few cases of MALT lymphoma, histiocytic, follicular, T-cell and others have also been described2, 10, 11). Hepatocellular carcinoma and metastasis from gastrointestinal (mostly colon) carcinoma are similar to PHL and are more common than. Normal levels of the tumor markers alpha-fetoprotein and carcinoembryonic antigen (CEA) are found in almost 100% of patients with PHL facilitating the differential diagnosis2, 10, 11). It is important to recognize that PHL may have a better prognosis than hepatocellular carcinoma or systemic disease that has secondarily affected the liver.
Since 1979 there have been 14 cases of PHL including 1 case of MALT lymphoma in Korea3-9). The age of patients on admission ranged from 18 to 78 years (median age 41 years) with no male to female preponderance (M:F=1:1). DLBL was also the most frequent histologic type of PHL in Korea (36%)39).
Because this disorder is rare optimal management and treatment modalities for PHL are not yet well-established. Surgical treatment, radiotherapy and chemotherapy have all been reported as treatment modalities alone or in combination2). Page et al. reported an excellent response rate associated with the use of multimodal chemotherapy alone (complete remission rate of 83.3%, 5-year relapse free survival rate of 83.1%)11). Contrary to Page et al. Scoazec et al. observed an inferior outcome in patients treated with chemotherapy alone compared to patients who were treated with surgery alone or chemotherapy plus radiotherapy modalities12). However, as Lei et al. pointed out, these discrepancies are attributable to patient selection. Chemotherapy is used mainly in patients who are not candidates for surgery or who had unresectable tumors, diffuse hepatic involvement, extrahepatic disease in the spleen or bone marrow, or high-grade histologic subtypes10). The prognosis for PHL may have been linked to the histologic subtypes, with short survival occurring in patients with unfavorable aggressive histologies2). Therefore, histologic subtypes and stage should be considered in the selection of the proper treatment modality for patients with PHL.
To our knowledge, there have only been two cases available with information on the treatment and response for PHL of MALT type3, 13). A female patient with MALT lymphoma (3 cm diameter of mass) was treated by segmental resection alone and was alive and well 12 months after diagnosis3). Another female patient with a MALT lymphoma (4 cm diameter of mass) was treated surgically followed by localized radiotherapy and was alive and well free of disease 2.5 years after the diagnosis13). We chose radiotherapy for our patient with a primary hepatic MALT lymphoma (5.7 4.8 cm-sized mass) because of the low grade histologic subtype and localized disease. Our case had 5 years of disease free survival. The patient had undergone radiotherapy with a total dose of 4,140 cGy and lived for 6 years. The doses of the radiotherapy used by Maes et al. were not reported13).
Previously, the primary therapeutic option for gastric MALT lymphoma was gastrectomy. As low-grade MALT lymphomas are very sensitive to radiation and respond well to lowdose radiation, radiotherapy is often used for the treatment of the gastric MALT lymphoma in order to preserve the stomach14). A recent report shows a 100% complete response rate with radiotherapy alone (median dose of 3,000 cGy) in 17 gastric MALT lymphoma patients15). Given the radiotherapy results for the gastric MALT lymphoma, radiotherapy alone may be a sufficient treatment modality for the treatment of primary hepatic MALT lymphoma.
In conclusion, it is suggested that radiotherapy should be considered as the preferred treatment for localized primary hepatic extranodal marginal zone B-cell lymphoma of MALT.



 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





