


INTRODUCTION
Rheumatoid arthritis (RA) is a progressive inflammatory disease with severe symptoms of pain and stiffness. RA is relatively common with its prevalence ranging from 0.5% to 1% of adults around the world [1]. Chronic persistent inflammation of RA often leads to joint destruction, deformity and limitation of function, which ultimately results in significant deterioration of quality of life (QoL). In addition to impairment of QoL, RA also shortens survival in most advanced patients, with systemic features such as fatigue, low-grade fever, and elevation of acute phase reactant. RA is characterized pathogenetically by immunologically driven, chronic synovitis, and production of autoantibodies, such as rheumatoid factor (RF) and anti-cyclic citrullinated peptide (CCP) antibodies [2]. Although the cause of RA is yet unknown, advances in the molecular biology led to in-depth understanding of its pathogenesis, and have fostered the recent development of novel treatments. The last decade has seen the dramatic change in the landscape of RA treatment with more aggressive therapy early in the disease course and with treatment guided by a structured assessment of disease activity, with the ultimate goal of reaching remission. In this review, recent understanding in the pathogenesis of RA is reviewed followed by the examples of therapeutics developed based on these understandings. Next, the advances in early diagnosis of RA and measurement of disease activity and its implication in the improvement of treatment outcome are discussed.
PATHOGENESIS OF RA
The pathogenesis of RA is not completely understood but the knowledge about pathobiology underlying the arthritis has been significantly increased in the last decade. Genetic susceptibility and environmental triggers were suggested by numerous studies. Adaptive and innate immune systems are both involved in the propagation of the disease. New vessel formation occurs in synovial tissue and leukocytes transmigrate into synovial compartment in early phase of RA. The cell migration is induced by the increased expression of adhesion molecules and chemokines [3,4]. T cells are abundant in synovial tissues of RA and T cell activation by antigen presenting cells (APCs) along with co-stimulation is essential in active synovitis. RA is considered to be a T helper 1 cell (Th1) type disease, but the role of Th17 has been increasingly emphasized. Th17 cells are subsets of T cells that produce interleukin (IL)-17A, 17F, 21, 22, and tumor necrosis factor (TNF)-╬▒ [5,6]. The pro-inflammatory cytokine, IL-1╬▓, 6, 21, and 23 induces the Th17 differentiation and suppress the activation of regulatory T (Treg) cells [7]. This imbalance between Th17 cells and Treg cells are important in T cell homeostasis towards inflammation [8].
To maximize T cell response, second signals called co-stimulatory signals are generally required. The co-stimulatory molecules CD28 and CD40 ligand are highly expressed on synovial T cells. CD28 on T cells binds with CD80 and CD86 on APCs, subsequently transmits a stimulatory signals with antigens presented by APCs [9]. Presentation of antigen to T cells by APCs without co-stimulation leads to anergy and apoptosis of poorly stimulated T cells. Cytotoxic T-lymphocyte antigen 4 (CTLA4) also binds with CD80 and CD86, which subsequently transmits inhibitory signal to T cells. The fusion protein CTLA4-Ig (abatacept), which competitively inhibits the CD28-CD80/86 co-stimulation showed treatment efficacy in RA [10].
The role of B cells in RA pathogenesis recently were being highlighted because the CD20+ B cell depleting therapy with rituximab showed beneficial effect in RA [11]. B cells may play several important roles in RA; antigen presentation and production of cytokines and RFs. Treatment with rituximab does not always decreases the level of autoantibodies of RA, which suggests the roles of B cells are not directly associated with autoantibody production but are mainly associated with adaptive immune response mediated by cytokines and interaction with T cells.
Pro-inflammatory cytokines play key roles in RA pathogenesis. They contribute to the underlying immune dysfunction and to immune-mediated target organ damages. TNF-╬▒ plays fundamental roles through promotion of angiogenesis, enhancing proliferation of T cells and B cells, inducing other inflammatory cytokines and chemokines and suppression of Treg cells [12]. TNF blockade is now widely used treatment for RA with high treatment efficiency. IL-6 also has pleiotropic roles in RA such as stimulating antibody production by B cells, differentiation of Th1 and Th17 cells and promoting acute phase responses and anemia [13,14]. The blockade of IL-6 receptor also showed successful results in treating RA patients [15]. IL-17 has been implicated in bone and cartilage damages through induction of matrix metalloproteinases (MMPs) and osteoclasts, as well as inhibition of proteoglycan synthesis [16]. Increased level of IL-17 in the synovium may be associated with increased risk of radiographic progression [17]. Recently, an anti-IL-17A antibody demonstrated modest efficacy in patients with RA [18].
The meshenchymal-derived, fibroblast-like synoviocytes (FLSs) also plays an important role in pathogenesis of RA. FLS from RA patients expresses high level of cytokines, chemokines, adhesion molecules, MMPs, and tissue inhibitors of metalloproteinase. They promote sustained T cell and B cell activation and involve in cartilage and bone destruction [19,20].
Bone erosion occurs in RA in response to prolonged, increased inflammation. At site of active RA, there is a dramatic imbalance between bone resorption and bone formation [21]. The receptor activator of nuclear factor kappa B ligand (RANKL) promotes osteoclast differentiation and bone resorption [22]. TNF-╬▒, IL-1, IL-6, and IL-17 induce osteoclastogenesis and thereby enhance bone erosion. Blockade of these molecules showed inhibitory effects on bone erosion in RA [23].
NEWLY DEVELOPED TREATMENTS FOR RA
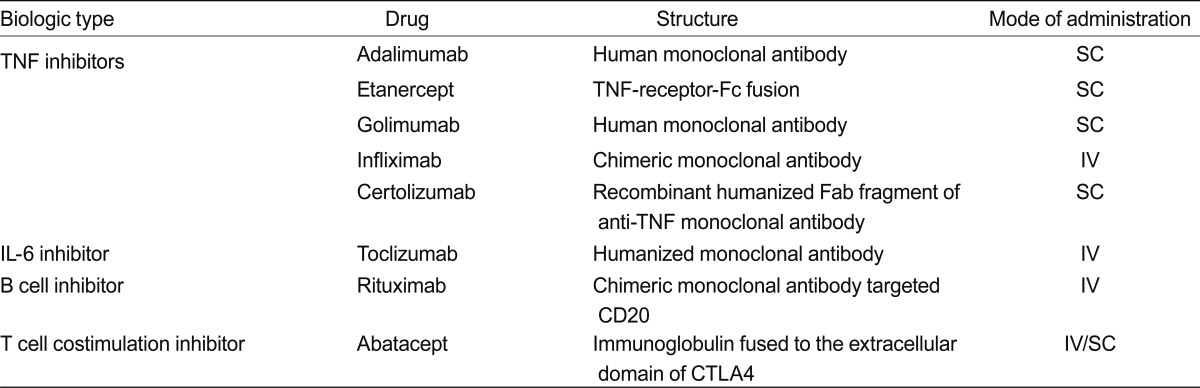
The advances in understanding the pathogenesis of RA have promoted the new therapeutic agents with improved treatment outcome (Table 1). Four classes of biologics are currently available; TNF inhibitors, B-cell inhibitors, T-cell co-stimulation inhibitors, and IL-6 inhibitors. IL-1 inhibitor, anakinra was developed for treatment of RA but it is now rarely used because of its relative weak efficacy. Recognition of new molecules that perform significant role in inflammatory process in RA has led to the development of new targeted agents.
TNF inhibitors were the first established biologic agents with successful translation from bench to bedside. Three TNF inhibitors; infliximab, etanercept, and adalimumab are currently used in Korea in clinical practice. Two additional TNF inhibitors, golimumab, and certolizumab have demonstrated acceptable safety and tolerability profile as well as efficacy comparable to 1st line TNF inhibitors in clinical trials [24]. Golimumab is human immunoglobulin G1 monoclonal antibody (mAb), administered subcutaneously at 4-week-intervals; certolizumab is a polyethylene glycol subcutaneous agent given every 2 weeks.
Tocilizumab is a humanized mAb against IL-6 receptor. Intravenous preparation given 4-weekly was licensed for RA, and a new subcutaneous formulation was recently evaluated in an open label study [25].
Abatacept (CTLA4-Ig) acts as a competitive inhibitor of the CD28-CD80/86 co-stimulatory interaction, which results in suppression of T cell activation. Abatacept showed similar response rate with other biologics [26]. It can be administrated both intravenously and subcutaneously. Different dosing schedules were established for different mode of administration [27].
Rituximab is a genetically engineered chimeric mAb targeting B cells bearing CD20 [10]. It was initially introduced for the treatment of lymphoma, and its treatment efficacy in RA was subsequently revealed. It is intravenously administrated as two 1,000 mg separated by 2 weeks. It is generally recommended for the patients who show inadequate response to TNF inhibitors or other biologics.
Besides the licensed biologics mentioned above, new drugs targeting other molecules have been proposed. Agents targeting various cytokines; anti-IL-7, 15, 17, 18, 21, 23, 32, and 33 were introduced [23]. Fully humanized IL-17 mAb, secukinumab were underwent phase II clinical trials with successful result [28]. Treatments targeting RANKL, Treg cells, granulocyte-macrophage colony-stimulating factor (CSF), and macrophage CSF are now under investigation [23,27].
Most promising results came from drugs targeting intracellular signaling molecules and transcription factors. Drugs targeting these small molecules can be administered orally, which may assure convenience and cost-effectiveness compared to parenteral biologics. Agents targeting Janus kinase (JAK) and spleen tyrosine kinase (Syk) showed good data in clinical trials. Tofacitinib, a selective JAK inhibitor showed significant efficacy and safety profiles in patients with active RA when used alone [28] or in combination with methotrexate (MTX) [29] in phase II trials. Phase III trials are undergoing with some early positive results [30]. Oral Syk inhibitor showed significant response compared to placebo in patients with active RA despite methotrexate therapy [31].
DIAGNOSIS
Because of the recent trends in early aggressive treatment of RA, the importance of accurate early diagnosis cannot be overestimated. Because of the lack of pathognomonic laboratory finding, the diagnosis of RA remains clinical one until now, however. The classification criteria defined by American College of Rheumatology (ACR, formerly American Rheumatism Association) (Table 2) has been widely used in the clinic until recently [32], and obviously the criteria include an item that should not be present in RA patients treated with current optimal therapeutics, such as bony erosion. Thus, these criteria are not helpful in achieving the goal of identifying patients who would benefit from early aggressive treatment. A joint working group of the ACR and the European League Against Rheumatism (EULAR) was formed to develop a new approach for classification of RA [1]. The 2010 ACR/EULAR classification criteria for RA was developed with the specific aim of facilitating the study of RA patients at earlier stages of the disease (Table 3).
It is important to stress that the new criteria should be applied only to eligible patients, in whom the presence of clinical synovitis, manifested as swollen joint in at least one joint, is present, and not to those with mere arthralgia or normal subjects. In addition, the use of the new criteria should be limited to subjects in whom there is no other diagnosis for their synovitis. Compared to previous criteria, symmetry is not a feature of the new criteria, although the higher the number of joints involved the greater the likelihood of symmetrical involvement. Because a detailed literature review showed that anti-CCP (ACPA) positivity did not add importantly to the discriminative value to classify an individual as having RA, beyond the information provided by RF when it is positive, the new criteria included both markers (ACPA and RF) equally in the criteria [2,33]. Validation in three of the cohorts available to the working group showed that the criteria were satisfied in 87% to 97% of the patients in whom the physicians chose to initiate methotrexate treatment [2]. The new criteria redefine RA, with a specific emphasis on identifying patients with a relatively short duration of symptoms who may benefit from early institution of disease modifying antirheumatic drug (DMARD) therapy or entry into clinical trials of promising new agents, and reflect hope that in the future, RA will no longer be characterised by erosive joint disease and persistence of symptoms [2].
EVALUATION OF DISEASE ACTIVITY
The primary consequences of RA are the structural joint damages leading to impairment of QoL. Over the past decade, the availability of effective biological therapies has drawn attention to the need for rheumatologists to accurately assess RA disease activity. The current treatment paradigm of RA has dramatically changed from classical approaches such as sequential monotherapy to early combination DMARDs. Recent treatment goal of RA is modeled upon that of other chronic diseases, such as hypertension and diabetes mellitus (DM), with tight control of blood pressure or blood glucose level to minimize long-term detrimental complication. The recent consensus of an international group of experts recommends a 'treat to target (T-2-T)' approach for RA treatment based on objective measures of disease activity and aims towards promptly achieving remission or a low disease activity [34,35]. This approach involves objective measurement of RA disease activity to direct treatment, and recommends that a target of disease activity be set for each individual patient. The ideal target for the treatment of all patients with RA is clinical remission, defined as the absence of signs and symptoms of inflammation. Disease activity has been shown to be the strongest predictor of disability in established RA, and remission is associated with less disability and improved QoL in comparison to even low disease activity states [36]. Because inflammation is the primary process that leads to joint damages, inflammatory changes are the main target of therapeutic interventions. Joint inflammation manifests in diverse ways, ranging from clinical symptoms and signs, such as pain, tenderness, swelling, limitation of range of motion to systemic manifestations, such as fatigue, mild fever, an increase in acute phase reactant and anemia of chronic disease. Each parameter is associated with different outcome variables, which makes the assessment of disease activity in RA very different from that of other chronic disease. For example, end-organ damage in DM or hypertension can be mostly averted by controlling a single parameter, such as blood glucose level and blood pressure. However, in RA, tender joint count is related with the level of actual disability, whereas swollen joint count is related to joint destruction and long-term outcome [37]. Erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level are also associated with joint damage and disability. Therefore, composite measures of disease activity are needed to portray disease activity in RA patients properly. Various definitions of disease activity have thus far been proposed, including ACR criteria, disease activity score (DAS), DAS28, simplified disease activity index (SDAI) and clinical disease activity index (CDAI). The core set for ACR response criteria include seven clinical end points: swollen joint count, tender joint count, physician's assessment of disease activity, patient's assessment of disease activity, patient's assessment of pain, and patient's assessment of physical function, and levels of an acute-phase reactant (either the CRP level or the ESR) [38]. An ACR20 response is defined as at least 20% improvement in both the tender joint count and the swollen joint count and at least 20% improvement in three of the five other core set measures.
The caveats of ACR20 response are as follows. First, recent therapeutic advance made 20% improvement less optimal, and other thresholds such as the ACR50 and the ACR70 were proposed. Second, the ACR20 is a dichotomous measure (i.e., the presence or absence of response), while information about relative improvement would be more useful to compare treatments. The DAS and DAS28 are continuous numerical measures that include tender and swollen joint count, patient's subjective assessment of disease activity, and a laboratory measurement of an acute phase reactant (Table 4). The original DAS uses the Ritchie articular index to enumerate tenderness in 53 joints and swelling in 44 joints [39]. The DAS28 employs a simplified 28-joint count that includes only the shoulders, elbows, wrists, metacarpophalangeal and proximal interphalangeal joints of the hands, thumb interphalangeal joints and knees and excludes the hips, ankles and feet. Although DAS < 1.6 was reported to correlate well with ACR remission criteria, and DAS28 < 2.6 with DAS < 1.6 [40-42], the implication of omitting foot and ankle joint in DAS28 is not well known. In our previous study, significant number of patients in clinical remission defined by DAS28 < 2.6 had foot metatarsophalangeal (MTP) and ankle involvement with some patients even showing isolated foot or MTP joint involvement without evidence of disease activity in other joints [38]. Among those who had radiograph available for examination, 30% showed radiographic joint damage at the time of clinical remission. Although a very well-designed recent report suggested that the inclusion of ankles and forefeet in the assessment of remission is not required, the study also showed that significant number of patients in remission had residual activity in the feet (26% by the Boolean definition and 36% by the SDAI definition) [43].
The SDAI and CDAI both use a 28-joint count to enumerate swollen and tender joints (Table 5) [44]. The SDAI consists of the numerical sum of the four components of the DAS28, with the addition of a physician global assessment of disease activity, while the CDAI omits the CRP, thereby allowing for its calculation at the time of the patient encounter without waiting for laboratory determination of an acute phase-reactant level [45].
TREATMENT
Classic treatment approach
In sequential monotherapy, treatment is initiated with traditional DMARD monotherapy, frequently MTX. If there is no response, monotherapy with another traditional DMARD is attempted [19]. This therapeutic approach is repeated several times until combination therapy with DMARDs, a biologic agent, or with a corticosteroid, is introduced as a last resort [20]. In the step-up approach, therapies with the least toxicity are utilized early, and more intensive therapies are added because of lack of response or toxicity [46]. DMARDs may be changed or added in patients with no response to treatment. Patients who continues to show a suboptimal response may receive additional DMARDs, either alone or in combination, or receive treatment with biologic agents [38]. These classic treatment approaches may be suboptimal for patients with RA, especially DMARD monotherapy. To prevent joint damage and ensuing disability, the inflammatory process in RA must be efficiently and rapidly controlled [47].
Clinical use of biologic therapy
TNF-blockers are predominantly the first biologics prescribed, with greatest experience and the largest amount of long-term follow-up data available for this drug class [39,48,49]. Approximately 20% to 30% of patients with established RA achieve clinical remission; whilst up to one-third of patients fail to obtain any significant benefit with TNF-blockers. Non-response may also manifest as a loss of response over time (secondary non-response). In patients with resistance or intolerance to initial TNF-blocker therapy, the choice of second line biologic is left to the discretion of the physician. Data from biologic registries suggest that patients who have failed two TNF-blockers are not likely to respond to a third [50]. In RF and anti-CCP negative patients, who are usually non-responsive to rituximab, the use of abatacept or tocilizumab may be recommended.
Korean T-2-T data
The response rate to non-biological DMARDs including MTX as well as biologic DMARDs has been scarcely reported among Asian RA patients. Since August 2010, DAS28 was measured every 3 months in every newly treated RA patients fulfilling 2010 ACR diagnostic criteria for RA who visited four Rheumatology Clinics of Hallym University Affiliated Hospitals [51]. One hundred and ninety patients were included (mean age, 54.5 years; female, 78.4%; mean disease duration, 15.0 months; RF positivity, 63.7%) between August 2010 and December 2011. Baseline DAS28 was 4.5. Corticosteroid was used in 89.0% of patients initially. DMARDs other than MTX prescribed included leflunomide in 14.7%, and sulfassalazine in 6.3%. At 3, 6, 9, and 12 months, 49%, 56.9%, 60.2%, and 73.2% of patients attained low disease activity. DAS28 remission was attained among 36.4%, 39.4%, 46.5%, and 57.1% during the same period. Health Assessment Questionnaire score decreased significantly from 1.06 at baseline to 0.45 at 6 months. After multivariate logistic regression analysis, the attainment of low disease activity at 3 and 6 months were only significantly associated with low baseline DAS 28. Our results show that current MTX based non-biological DMARD therapy induces low disease activity in a significant portion of Korean patients with RA and that prescription of MTX along with tight control of disease activity can improve patient outcome.
CONCLUSIONS
Recent guidelines suggest remission as the treatment goal for patients with RA. In addition, prevention and control of joint damage and improvement in QoL are important goals. To achieve these goals, a multidisciplinary approach to reduce disease activity with DMARDs and biological therapy is needed. The ultimate goal of therapy in RA would be clinical and radiographic remission, which is maintained after withdrawal of therapy. We also need to find ways to identify those patients who are at risk for more rapid disease progression who would benefit from intensive therapy early in the course of disease.




 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





