


INTRODUCTION
From embryonic development to adulthood, blood vessels play a fundamental physiological role in supplying oxygen and nutrients, removing catabolic waste, and circulating cells for immune surveillance [1,2].
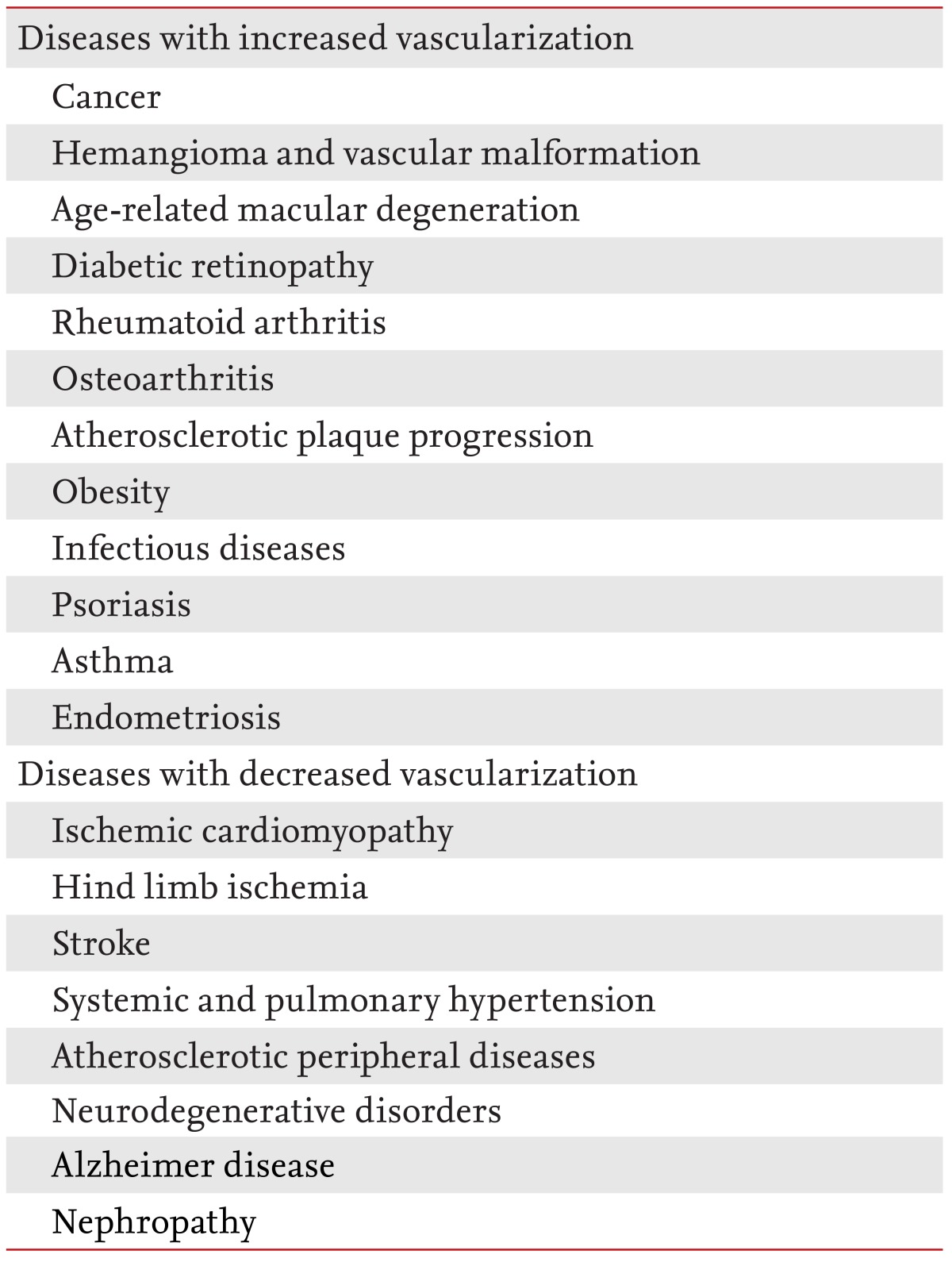
It is unsurprising that structural alterations or functional aberrations of vessels are involved in a plethora of diseases [3,4]. These diseases may be divided into two groups. The first involves inadequate vessel maintenance and growth; it includes diseases such as myocardial infarction, stroke, neurodegenerative or obesity-associated disorders, and requires proangiogenic therapy. The second involves disproportionate vascular growth and abnormal remodeling. This group includes cancer, inflammatory disorders, ophthalmic neovascular diseases, and requires antiangiogenic therapy (Table 1).
FACTORS DRIVING PHYSIOLOGICAL AND PATHOLOGICAL ANGIOGENESIS
The development of functional vessels by angiogenesis and arteriogenesis requires the cooperation of several growth factor families, their related receptors, multiple cell types, and the presence of certain conditions, such as hypoxia [9].
Understanding this process has allowed the identification of a large number of targets for the inhibition of angiogenesis. Some of these targets have been used for antiangiogenic therapy, whereas many others have the potential to become new validated targets. The following is a summary of the different activities of the molecule families that are active in angiogenesis.
The vascular endothelial growth factor (VEGF) family
VEGF (also known as VEGF-A) is the main member of the VEGF family, and plays a major role in angiogenesis. Its activity is exerted through the binding of two receptors: VEGF receptor 1 (VEGFR-1; also known as Flt-1) and VEGFR-2 (also known as KDR or Flk-1). The latter plays a main role in endothelial activation in conjunction with neuropilin (NRP) receptors 1 and 2 that act as coreceptors to enhance the activity of VEGFR-2 [10]. The soluble isoforms of VEGF stimulate vessel enlargement, whereas the isoforms that bind to the extracellular matrix promote vessel branching [11,12]. VEGF produced by endothelial cells maintains vascular homeostasis.
VEGF-C is a ligand of the VEGFR-2 and VEGFR-3 receptors. It plays an important role in stimulating endothelial cells to express the tip cell phenotype. These endothelial cells become motile, invasive, and protrude filopodia, which drives new vessel formation [13]. VEGFR-3 plays a role in vascular formation during early embryogenesis. Later, it becomes a key regulator of lymphangiogenesis or the formation of new lymphatic vessels from pre-existing ones [14].
Placental growth factor (PlGF) is relevant only in pathological conditions [15-17]. The activation of its specific receptor, VEGFR-1, directly or indirectly stimulates angiogenesis. PlGF is able to recruit and stimulate bone marrow-derived endothelial progenitor and myeloid cells needed to sustain the angiogenic process [18]. PlGF contributes to the unequal polarization of tumor-associated macrophages (TAMs) between the M1 and M2 phenotypes [19].
Like PlGF, VEGF-B is not required for physiological angiogenesis and it specifically recognizes VEGFR-1. Its angiogenic activity is limited to certain tissues such as the heart [20]. Interestingly, PlGF and VEGF-B can stimulate the growth of new vessels without inducing adverse effects such as increased permeability or leakage, as observed in diverse preclinical models [21,22].
VEGFR-1 remains the most elusive in terms of angiogenic function. This is most likely due to the fact that it is expressed in different cell types and it is activated by three members of the VEGF family: VEGF-A, VEGF-B, and PlGF [23]. Based on its weak tyrosine kinase activity, VEGFR-1 has been defined as a decoy receptor for VEGF, and determines the amount of free VEGF available to activate VEGFR-2. This explains why VEGFR-1 loss results in vessel overgrowth [24]. Alternatively, VEGFR-1 activation on angiogenic endothelial, stromal, and myeloid cells stimulates pathological angiogenesis [25].
The platelet derived growth factor (PDGF) family
The maturation of new blood vessels by arteriogenesis occurs through the covering of nascent vessels by mural cells. The PDGF family contributes to this process along with angiopoietins (ANGs) and transforming growth factor β (TGF-β) [26]. PDGF-B is produced by endothelial cells to recruit pericytes from the local environment or perivascular pericyte progenitors from the bone marrow [27,28] that express PDGF receptor β. In turn, pericytes produce VEGF, which is essential for endothelial survival and protects vessels from VEGF blockade.
Other members of this family also have angiogenic functions. PDGF-CC stimulates vessel growth and maturation, but attenuates the response to anti-VEGF treatment [29]. Inhibition of PDGF-DD suppresses ocular neovascularization, whereas PDGF-DD overexpression normalizes tumor vessels and improves drug delivery.
The fibroblast growth factor (FGF) family
Basic FGF was one of the first angiogenic factors to be discovered. Other members of this family, such as FGF1 and FGF9, have angiogenic and arteriogenic properties. The activation of FGF receptors (FGFRs), on endothelial cells or other cell types, sustains angiogenesis directly or by inducing the release of angiogenic factors. This has been confirmed by data indicating that FGF signaling promotes tumor angiogenesis [30].
Low levels of FGF are needed to maintain vascular integrity because inhibition of FGFR signaling in quiescent endothelial cells causes vessel disintegration [31].
The ANG and tyrosine kinase endothelial (TIE) family
This family plays a crucial role in the maintenance of vessel quiescence but vessels must be able to respond to angiogenic stimuli [32].
The two main ligands of the ANG family, ANG1 and ANG2, have the same target receptor, TIE2. ANG2 acts as a competitive antagonist of ANG-1. A second receptor, TIE1, exists but a ligand has not yet been identified. The function of this orphan receptor remains elusive [32].
ANG1 stimulates TIE2 clustering at cell-cell junctions, which sustains vessel quiescence and promotes mural cell coverage of vessels [33]. In the presence of angiogenic stimuli, activated endothelial cells produce ANG2 that antagonizes ANG1/TIE2 signaling enhancing mural cell detachment, vascular permeability, and endothelial cell sprouting. ANG2 also promotes angiogenesis by recruiting proangiogenic TIE2-expressing monocytes (TEMs) [34].
The cadherin family
In quiescent vessels, the interconnections between endothelial cells are crucial to form a continuous monolayer. This formation has been described as a cellular phalanx. Vascular endothelial cadherin (VE-cadherin) is vital for endothelial cell-cell adhesion. VE-cadherin also promotes vessel stabilization by inhibiting VEGFR-2 signaling and activating TGFR pathways [35]. VE-cadherin is strictly regulated by oxygen concentration in a feedback loop so that vessel perfusion may be optimized depending on the oxygen supply [36]. A reduction in VE-cadherin between adjacent cells by endocytosis occurs in response to angiogenic stimuli and supports new vessel formation. The localization of VE-cadherin in the filopodia of tip cells allows the establishment of new connections with other sprouting vessels.
The notch-and delta-like ligand 4 (DLL4) signaling
During new vessel formation, the tip cells migrate toward the angiogenic stimuli, while the neighboring endothelial cells (stalk cells) proliferate [8]. The activation of VEGFR-2 by VEGF in tip cells upregulates DLL4 expression, which determines the activation of NOTCH in stalk cells [37]. There is downregulation of VEGFR-2 and upregulation of VEGFR-1 in stalk cells, which become less responsive to VEGF as opposed to other VEGF family members able to bind VEGFR1. This demonstrates a complexity of endothelial cells by activation of a fine-tuning of VEGFR-1 and VEGFR-2 expression in response to neighboring stimuli that determine the tip or stalk functional phenotypes [38].
The ephrin (EPH) family
EPH receptors and ligands modulate cell contact dependent patterns. During vasculogenesis, arterial and venous territories are specified by EPH-B2 and EPHB4 expression, respectively. Their interactions avoid vessel repulsion while also separating the respective vessel territories. EPH-B2 also promotes the recruitment of mural cells and bone marrow-derived endothelial progenitor cells [39,40].
The semaphorin (SEMA) family
The SEMA family is composed of a large number of secreted or Gpi-anchored proteins that are able to bind plexin receptors or NRP coreceptors [41]. Many SEMAs inhibit tumor angiogenesis (SEMA3A, SEMA3B, SEMA3D, SEMA3F, and SEMA4A), whereas SEMA3C and SEMA4D promote tumor angiogenesis. The loss of the plexin-d1 receptor in mice induces erroneous vessel navigation, because the endothelial cells cannot recognize the repulsive SEMA3E signals.
APPROVED ANTIANGIOGENIC DRUGS FOR CANCER THERAPY AND AGE-RELATED MACULAR DEGENERATION
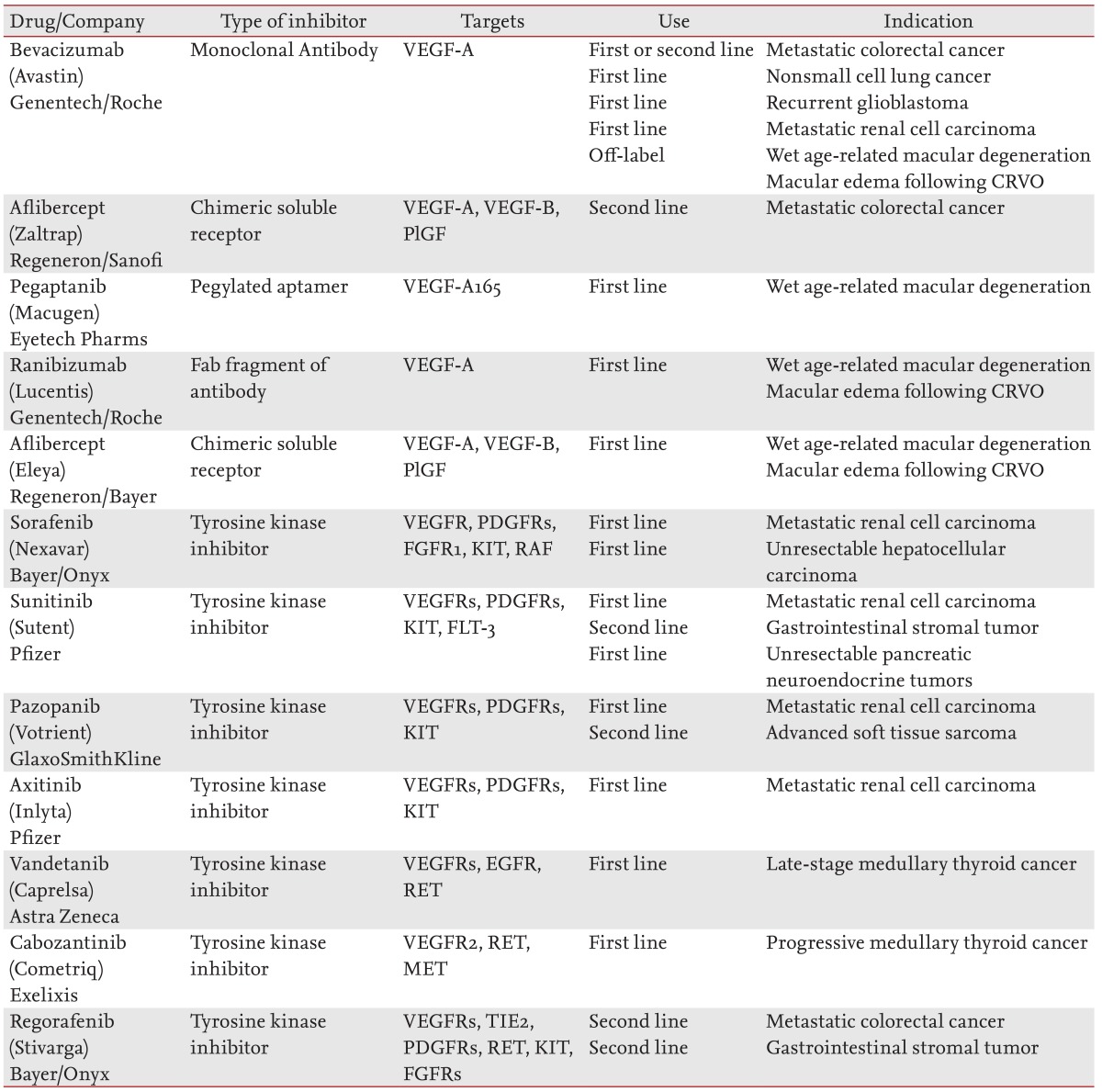
Despite the large number of putative targets for antiangiogenesis therapy and multiple reported compounds with antiangiogenic activity [5,9], the vast majority have failed despite encouraging preclinical results [7,42]. The exceptions are the drugs that target VEGF ligands or receptors [10]. Most of the antiangiogenic agents that entered the drug development pipeline in the past decade target VEGF signaling pathways. However, efforts using other targets such as ANGs and integrins are ongoing. The approved antiangiogenic drugs treat various tumors or age-related macular degeneration (AMD) (Table 2).
Bevacizumab (Avastin, Genentech, San Francisco, CA, USA), a humanized anti-VEGF-A monoclonal antibody [43], was the first antiangiogenic drug approved by the U.S. Food and Drug Administration. The clinical trial was a randomized double-blind phase-III study in which bevacizumab was administered in combination with a bolus of irinotecan, 5-fluorouracil, leucovorin chemotherapy as a first-line therapy for previously untreated metastatic colorectal cancer (mCRC). The median survival and progression-free survival times were increased with the addition of bevacizumab [44].
Currently, bevacizumab is approved for first-and second-line treatment of mCRC in conjunction with 5-fluorouracil-irinotecan or 5-fluorouracil-oxaliplatin-based chemotherapy. It is also approved for first-line treatment of advanced nonsquamous nonsmall cell lung cancer in combination with carboplatin and paclitaxel in patients who have not yet received chemotherapy. More recently, it has been approved as a single agent in adult patients with recurrent glioblastoma multiforme who had tumor progression after initial treatment, and in conjunction with interferon Îą to treat metastatic renal cell carcinoma (RCC) [45].
Although bevacizumab is generally well tolerated, some serious and unusual toxicities were observed, including: gastrointestinal perforation, hemorrhage, proteinuria, venous or arterial thromboembolic events, and hypertension. The most common adverse events were asthenia, abdominal pain, headache, diarrhea, nausea, and vomiting [6].
Aflibercept (Zaltrap, Regeneron and Sanofi Aventis, Bridgewater, NJ, USA), previously known as VEGF-trap, is a recombinant fusion protein that consists of VEGF-binding portions from the extracellular domains of human VEGFR 1 and 2 fused to the Fc portion of human immunoglobulin G1. It functions as a decoy receptor for proangiogenic members of the VEGF family: VEGF-A, VEGF-B, and PlGF. It has been approved for second-line treatment of mCRC that is resistant or has progressed following treatment with an oxaliplatin-containing regimen. It is used in combination with 5-fluorouracil, leucovorin, and irinotecan (FOLFIRI) [46].
The adverse effects were similar to those observed with bevacizumab, but common adverse reactions such as neutropenia, diarrhea, hypertension, leukopenia, stomatitis, fatigue, proteinuria, and asthenia, were reported more frequently with those treated with aflibercept and FOLFIRI.
Several multitargeted tyrosine kinase inhibitors (TKIs), which block the signaling of pathways such as VEGF, have been approved for cancer therapy. Sorafenib (Nexavar, Bayer HealthCare Pharmaceuticals, West Haven, CT, USA) targets VEGFRs, PDGFRs, FGFR1, Kit, and Raf kinases (mainly C-Raf as opposed to B-Raf). It was approved for first-line treatment of metastatic RCC in 2005 [47] and for the treatment of unresectable hepatocellular carcinoma in 2007 [48].
Sunitinib (Sutent, Pfizer Inc., New York, NY, USA) inhibits the tyrosine phosphorylation of VEGFRs, PDGFRs, c-kit, and Flt-3, and was approved for first line treatment of metastatic RCC in 2006 [49]. It was also approved for the treatment of gastrointestinal stromal tumors (GIST) following progression during treatment with imatinib (Gleevec, Novartis, Basel, Switzerland) or with intolerance to the drug [50]. More recently (May 2011), sunitinib has been approved for the treatment of pancreatic neuroendocrine tumors in patients with unresectable, locally advanced, or metastatic disease [51].
Pazopanib (Votrient, GlaxoSmithKline, Stevenage, UK) targets VEGFRs, PDGFRs, and c-kit, and was approved in 2009 for first-line treatment of metastatic RCC [52]. In April 2012, it was also approved for the treatment of advanced soft tissue sarcoma in patients who had received prior chemotherapy [53]. Axitinib (Inlyta, Pfizer Inc.) has the same targets as Pazopanib and was approved in January 2012 for the treatment of advanced RCC that progressed despite first-line therapy [54].
Vandetanib (Caprelsa, AstraZeneca, Wilmington, DE, USA) inhibits VEGFRs, EGFR, and RET-tyrosine kinase. It was approved in April 2011 for the treatment of symptomatic or progressive medullary thyroid cancer (MTC) in patients with unresectable, locally advanced, or metastatic disease [55].
Cabozantinib (Cometriq, Exselixis Inc., San Francisco, CA, USA) inhibits the activity of multiple tyrosine kinases, including RET, MET, and VEGFR2. It was approved in November 2012 for the first-line treatment of patients with progressive metastatic MTC [56].
Regorafenib (Stivarga, Bayer HealthCare) was approved in September 2012 for second-line treatment of patients with mCRC who have been previously been treated with 5-fluorouracil, oxaliplatin, and irinotecan based chemotherapy, with an anti-VEGF therapy, and, if KRAS wild type, with an anti-EGFR therapy [57]. In February 2013, it was also approved for the second-line treatment of patients with advanced GIST that cannot be surgically resected and no longer responds to imatinib and sunitinib [58]. Regorafenib and its active metabolites inhibit multiple membrane-bound and intracellular kinases that are involved in normal cellular functions and pathologic processes, including: RET, VEGFRs, KIT, PDGFRs, FGFRs, TIE2, and Raf.
The listed TKIs have many adverse effects; the most common include: fatigue, diarrhea, nausea, and vomiting. Hypertension, as with bevacizumab and aflibercept, also occurs with TKIs, and often requires medical intervention with standard antihypertensive therapy. Hypertension developed in 11% of patients receiving bevacizumab and is now recognized as a standard side effect of VEGF blockers [6].
Three anti-VEGF drugs are used for the treatment of the wet (neovascular) form of AMD. AMD causes blindness due to the formation of leaky neovessels [59].
The first, Pegaptanib (Macugen, Eyetech, New York, NY, USA), is a pegylated, 28-base ribonucleic aptamer that specifically targets the VEGF165 isoform that was approved in 2004. Macugen slowed the progression of vision loss, but did not yield clinically significant improvements in visual acuity [60].
The second drug, ranibizumab (Lucentis, Genentech), is a recombinant, humanized monoclonal antibody Fab fragment derived from bevacizumab that was approved in 2006. Phase 3 clinical studies demonstrated that monthly intravitreal injections of ranibizumab prevented the loss of visual acuity in ~95% of patients and improved visual acuity in 25% to 33% of patients during a 24-month treatment period [60,61]. Ocular adverse events, such as endophthalmitis or uveitis, attributable to either the injection procedure (intravitreal) or Ranibizumab were observed in 1% to 2% of patients during the 2-year treatment period. With respect to the potential systemic anti-VEGF side effects, the rates of hypertension were not imbalanced, and adverse events associated with proteinuria were not reported. Ranibizumab has been also approved for the treatment of macular edema following central retinal vein occlusion (CRVO) [62].
Ophthalmologists have begun to use off label Bevacizumab instead of Ranibizumab due to the enormous difference in terms of cost and reports of similar beneficial effects [63].
In November 2011, Aflibercept (Eylea, Regeneron, Tarrytown, NY, USA) has also been approved for the treatment of patients with AMD and macular edema following CRVO [64].
Since VEGF plays an important role in the physiological homeostasis of the retina [65], important side effects have been reported with respect to retinal function [66,67]. However, several clinical trials with Ranibizumab, Bevacizumab, and Aflibercept are ongoing to explore the treatment of neovascular ocular pathologies, such as diabetic retinopathy and retinopathy of prematurity [68,69].
ANTIANGIOGENIC DRUGS IN CLINICAL DEVELOPMENT
Tens of clinical trials are ongoing for new antiangiogenic agents, many of which are receptor TKIs. Please see http://www.cancer.gov/clinicaltrials or http://clinicaltrials.gov/ct2/home for more information. For other agents, it is interesting to note drugs directed towards specific targets that are not related to the mechanism of action of currently approved drugs.
Trebanabib (Amgen, Thousand Oaks, CA, USA), a peptide fused to the Fc portion of an antibody (peptibody), potently and selectively inhibits ANG1 and ANG2 binding to the TIE2 tyrosine kinase receptor [70]. It is currently in phase 3 clinical trials for the treatment of multiple tumor types.
Velociximab (PDL BioPharma, Fremont, CA, USA; and Biogen Idec, Cambridge, MA, USA), a chimeric monoclonal antibody that inhibits a5b1 integrin functional activity has passed phase 2 trials [71].
The first agent able to target integrins that has reached phase 3 of clinical development, Cilengitide (Merck KGaA, Darmstadt, Germany) [72], is a cyclic peptide that blocks av integrins. Unfortunately, this drug did not meet the primary endpoint in patients with newly diagnosed glioblastoma.
In addition, two promising human antibodies that also interfere with VEGF pathways, specifically targeting VEGFR-1 or VEGFR-2, are in clinical development. Ramucirumab (ImClone/Lilly, ImClone Systems, Branchburg, NJ, USA) blocks VEGFR-2 signaling and has recently met the primary endpoint of improved overall survival. It also prolonged the progression-free survival time in patients with metastatic gastric cancer [73]. Ramucirumab and Icrucumab (ImClone/Lilly), which blocks VEGFR-1 [74] are currently undergoing diverse phase 3 studies for multiple tumor types.
CONCLUSIONS
Despite great success in the development of several antiangiogenic drugs, the clinical outcomes in cancer patients treated with antiangiogenic therapy have indicated this strategy to be more challenging than anticipated [9,75-77].
Clinical benefit is reflected by the lengthening of progression-free survival time in advanced colorectal, lung, renal, pancreatic, neuroendocrine, and ovarian cancer and by the longer overall survival times in patients with metastatic colorectal and renal cancer. Antiangiogenic treatment generally prolongs the survival of responsive patients in the order of months, but the clinical benefit is not usually sustained or is decreased to absent with some types of cancer. Many cancer therapies have modest effects on overall survival times. Increases in overall survival were obtained in only 12% of 73 randomized phase 3 trials for bevacizumab, trastuzumab, and other targeted therapies in addition to a range of chemotherapeutic agents for metastatic breast cancer over the past 30 years [78].
Another limitation of antiangiogenic cancer treatment is evidenced by the results of both preclinical and clinical research into angiogenic mechanisms. Tumors can adapt to the presence of angiogenesis inhibitors so as to functionally evade the therapeutic blockade of angiogenesis [75]. In contrast to the traditional concept of drug resistance acquisition based on target gene mutation or alteration in drug uptake and efflux [79], evasive resistance for angiogenesis is essentially indirect [75]. VEGF blockade can aggravate hypoxia resulting in upregulation of angiogenic factors, such as PlGF, FGFs, chemokines, and EPHs, and also lead to recruitment of proangiogenic bone marrow-derived cells, including TEMs and TAMs. These responses may rescue tumor vascularization.
The observation that cancer cells can adapt by aggressively migrating into normal tissue after VEGF blockade [80,81] has opened an important debate on whether antiangiogenic treatment may lead to more invasive and metastatic tumors [82]. This raises new concerns that must be considered in future angiogenic therapy development.
Currently, the main concern is how antiangiogenic cancer therapy can be improved. An important focus will be the ability of drugs to recognize more angiogenic targets. The importance of this quality is evidenced by the fact that TKIs are effective as monotherapy since they act on several targets, while anti-VEGF antibodies or recombinant proteins require a combination with chemotherapy. Another consideration regards the dose and duration optimization for antiangiogenic drug delivery since these have not been clinically optimized [83]. Moreover, anti-VEGF agents could be combined with agents that target the escape pathways identified in clinical studies of ANG2, PlGF, stromal cell-derived growth factor 1Îą, and chemokine receptor type 4 [84].
The ability to prevent inflammatory cell recruitment [85] and tumor vessel normalization [86] should be explored in the future to improve anticancer immune therapy and prevent metastatic spread.
In contrast to cancer therapy outcomes, those obtained for AMD or CRVO have been more impressive. This may be due to the fact that the neovascularization associated with AMD is the primary cause for irreversible blindness in advanced countries. Anti-VEGF agents used as monotherapy prevented the loss of visual acuity in ~95% of patients, and improved visual acuity in 25% to 33%, which was never obtained with previous treatments [61,63]. Also for ocular therapy, the ability to block more targets involved in neovascular formation, particularly those associated with the immunological component of the disease [87], as well as preventing the numerous side effects, could represent a further advancement in therapeutic success.
In conclusion, the clinical data from the first decade of antiangiogenesis therapy along with the knowledge that pathological angiogenesis plays a role in a large number of complex diseases should result in further advances in angiogenic therapy.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





