


To the Editor,
Most congenital nephrogenic diabetes insipidus (NDI) is X-linked recessive, and < 10% of congenital NDI cases exhibit autosomal inheritance. Several cases of congenital NDI have been reported in Korea, most of which did not lead to azotemia. Although a few reports have shown that chronic kidney disease can be induced by recurrent dehydration and glomerular embolism, congenital NDI usually does not progress to renal failure if subjects are adequately hydrated. We report a case of NDI that progressed to end-stage renal disease (ESRD) with a brief review of the literature.
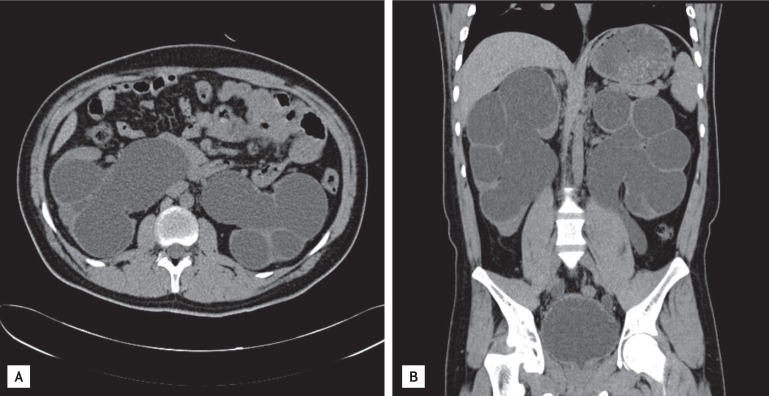
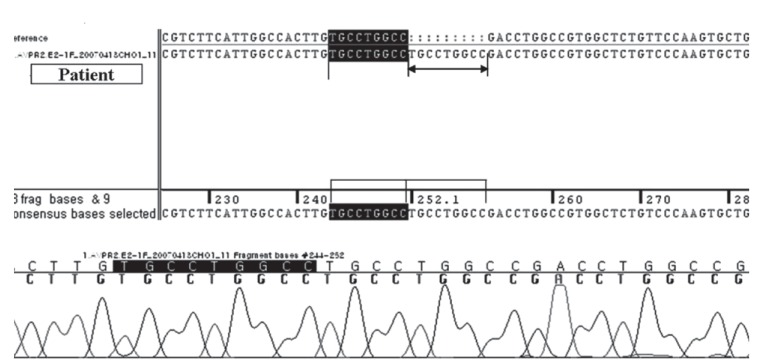
A 26-year-old male presented at a local clinic with a 3-month history of weight loss and a 6-month history of general malaise. He was transferred to our hospital with a high creatinine level. He was diagnosed with NDI at the age of 6 years and had been treated for 5 years before this presentation. He had received no treatment thereafter and developed 5 to 6 L polyuria per day. His older brother was healthy without such symptoms. There was no family history of the disease. His vital signs were stable, but a firm mass with an approximate diameter of 8 cm was noted on a physical examination of the abdomen. His laboratory findings were: blood urea nitrogen, 81.0 mg/dL and creatinine, 9.44 mg/L. Blood osmotic concentration was 329 mOsm/kg, and urine osmotic concentration was 170 mOsm/kg. The patient had developed metabolic acidosis; an arterial blood gas analysis revealed pH, 7.326; pCO2, 34.9 mmHg; pO2, 98 mmHg; and HCO3, 17.8 mmol/L. Urinalysis showed specific gravity, 1.015; protein, 3+; occult blood, 2+; and negative white blood cells. Urine volume on a 24-hour urine test was 6,500 mL, protein was 4,834.8 mg/day, and creatinine clearance was 11.74 mL/min, suggesting decreased renal function. Abdominal and pelvic computed tomography scans revealed bilateral atrophied kidneys and markedly dilated pelvis and calyces. The bladder was extended up to the umbilical level and was severely dilated (Fig. 1). No findings indicating a ureteral obstruction were detected. Genomic DNA was extracted, and an AVPR2 gene mutation was identified. An analysis of the base sequences in all exons and adjacent introns indicated a duplicate sequence of nine bases between no. 244 and 252 of exon 2 (Fig. 2). No AQP2 gene mutation was found. The patient is currently on hemodialysis three times per week, which was started during hospitalization due to aggravation of general malaise and the development of uremic symptoms, such as nausea and vomiting.
In most cases, congenital NDI is manifested by a variety of clinical features during infancy. Recurrent fever, vomiting, and irritability are manifested within 1 week of birth, and serious sequelae, such as retarded growth or mental disorders, may develop due to recurrent hypernatremia and dehydration. However, the increased thirst is managed by ingesting a large volume of water, which produces unconcentrated urine, leading to hydronephrosis, ureteral dilatation, or mega bladder without an anatomical ureteral obstruction in 15% to 67% of patients [1]. In our case, severe hydronephrosis, ureteral dilatation, and megabladder were observed. Differentiating congenital NDI from the acquired form can be difficult because both diseases are accompanied by polyuria and hydronephrosis. Therefore, a diagnostic methodology other than the clinical findings is mandatory to confirm congenital NDI. Gene analysis has become the best confirmative diagnostic method for congenital NDI. At present, > 150 AVPR2 gene mutations have been reported [2]. Most of these mutations are missense or nonsense AVPR2 or frameshift or inframe deletions. Cheong et al. [3] reported six mutations in the vasopressin V2 receptor gene causing NDI. Among these mutations, one was a short duplication of nine bases (bases 315 to 323), resulting in the insertion of three amino acids (Cys-Leu-Ala) in the transmembrane domain. They explained that the duplicated Cys residue was predicted to cause a conformational change because Cys residues form disulfide bonds. In our case, symptoms occurred only in the patient and not in his family members. Analysis of the base sequence revealed a duplicate sequence of nine bases between no. 244 and 252 of exon 2 (Cys-Leu-Ala). Our results were consistent with those of Cheong et al. [3]. However, we do not know whether the short duplication case of Cheong et al. [3] progressed to ESRD. Our patient was diagnosed with ESRD and currently undergoes hemodialysis treatment three times per week. Although chronic kidney disease rarely develops in a patient with congenital NDI if an adequate volume of water is supplied, it can occur due to recurrent dehydration, glomerular embolus, atrophy of renal parenchymal tissue, long-term hydronephrosis, or ureteral dilatation [4]. A case of NDI in Korea that was inherited as an autosomal dominant trait and subsequently progressed to renal failure was reported in 2004. That patient presented with polyuria and was subsequently diagnosed with congenital NDI as an adult [5]. In our case, the patient was diagnosed with congenital NDI at 6 years of age and intermittently received drugs such as thiazide. However, he did not receive any treatment after the age of 19 years. He was diagnosed with ESRD when he presented to our clinic with fatigue and general malaise.
In conclusion, evaluating at-risk infants by monitoring growth and periodically measuring serum sodium concentration to identify unrecognized hyperosmolality and early dehydration, as well as annual renal ultrasonography to monitor for hydronephrosis, are needed as early as possible to allow for a prompt diagnosis and treatment to reduce morbidity. In addition, genetic counseling and an evaluation of high-risk family members, such as prenatal testing, are highly recommended.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





