


INTRODUCTION
Idiopathic pulmonary arterial hypertension (IPAH) is a rare and incurable disease that is not associated with an identifiable cause or family history and is categorized as group 1.1 in the Nice classification system [1]. As pulmonary vascular resistance (PVR) and right heart failure progresses in patients with IPAH, several symptoms may present, including dyspnea, chest pain, fatigue, peripheral edema, and syncope. Without appropriate treatment, the median survival period in IPAH cases is 2.8 years [2]. Many studies have been conducted to determine the best management approach for IPAH. An important pathophysiological finding of IPAH is thrombotic arteriopathy, which influences disease development if it progresses [3]. Biomarkers associated with hypercoagulability, such as fibrinogen, tissue plasminogen activator, plasminogen activator inhibitor-1, and von Willebrand factor, also increase in patients with IPAH [3,4]. In accordance with these findings, anticoagulation therapy with warfarin improves survival of patients with IPAH [5-9]. A recent large prospective cohort study also demonstrated a survival benefit from anticoagulation in patients with IPAH [10].
The full benefits and role of anticoagulation in the treatment of IPAH remain uncertain; however, some reports have suggested that warfarin has limited survival benefit in cases of IPAH [11,12] but no prospective randomized controlled studies of this treatment have been performed. In addition, these earlier studies mostly originated from Western countries [5,6,8-12], and there is no comparable warfarin treatment study on Korean patients with IPAH. The purpose of this study was to evaluate the survival benefits of warfarin in an IPAH cohort from a Korean tertiary hospital.
METHODS
Study design and subjects
We performed a retrospective cohort study of patients > 20 years of age with a diagnosis of pulmonary arterial hypertension (PAH) made by one pulmonologist at a tertiary hospital in Korea between January 1994 and February 2013. To exclude patients with associated PAH, we reviewed the medical history (medications and previously diagnosed diseases) and test results (chest computed tomography [CT], ventilation/perfusion scan, echocardiography, liver ultrasonography, pulmonary function test [PFT], autoantibody test, liver function test, and human immunodeficiency virus test) for each patient. We then selected patients with IPAH who had undergone right heart catheterization (RHC) and excluded cases that did not meet the PAH diagnostic criteria, i.e., mean pulmonary artery pressure (PAP) > 25 mmHg, pulmonary capillary wedge pressure < 15 mmHg, and PVR > 3 Wood units at rest on RHC. We also excluded patients with IPAH who had positive vasoreactivity on RHC, which typically indicates a slower disease progression and a better prognosis than patients with a negative vasoreactivity test [6,13]. A positive vasoreactivity test is defined as a reduction in mean PAP Ōēź 10 mmHg and an absolute final mean PAp < 40 mmHg without a decrease in cardiac output (CO). We divided the patients with IPAH confirmed to have negative vasoreactivity by RHC into a ŌĆ£warfarin groupŌĆØ or a ŌĆ£non-warfarin groupŌĆØ based on taking warfarin within 1 year of the diagnosis. This study was approved by the Institutional Review Board of Asan Medical Center. Because this was a retrospective study, the requirement for informed consent was waived. We used unique identifier numbers to maintain patient confidentiality.
Outcome measures
We evaluated the survival effects of warfarin on IPAH by comparing patient survival periods in the warfarin and non-warfarin groups. We judged survival status of the patients using the hospital electronic medical records or Korean National Health Insurance data. The survival period was calculated from the day of diagnosis to death or the last hospital follow-up day, and was compared between groups using the Kaplan-Meier method and the log-rank test.
Baseline data collection
We collected clinical and hemodynamic data on the patients with IPAH at the initial diagnosis, including sex, age, body mass index, smoking history, time from onset of symptoms to diagnosis, symptoms at admission, World Health Organization (WHO) dyspnea functional class, laboratory findings, PFT, echocardiography, RHC, chest CT, and medications. Another pulmonologist reviewed all patient medical records, hemodynamic variables, and vessel diameter on chest CT at least twice. To improve accuracy, all data were rechecked by two other specialists in the respiratory division.
Pulmonary function test
We measured diffusing capacity (DLCO) routinely and performed a 6-minute walk test according to the American Thoracic Society guidelines [14]. DLCO was measured using a single breath technique and the Vmax ENCORE 22 (CareFusion Corp., San Diego, CA, USA). The measurements were compared to predicted values using reference equations from Park and the European Community for Steel and Coal [15,16].
Echocardiography
We evaluated ejection fraction (EF), left ventricular end-diastolic diameter (LVEDD), right ventricular end-diastolic diameter (RVEDD), and tricuspid regurgitation velocity by echocardiography. Echocardiography was usually performed using a Philips iE33 ultrasound system (Philips Healthcare, Andover, MA, USA). EF was measured by collating several methods and using the Quinones formula based on the parasternal views or using the quantitative two-dimensional biplane volumetric Simpson method from four- and two-chamber views [17,18]. LVEDD and RVEDD were measured using the two-dimensional linear or M-mode method from the parasternal long-axis view or the apical four-chamber view [18,19]. Systolic PAP was calculated using the trans-tricuspid gradient plus right atrial pressure (RAP), where the trans-tricuspid gradient is 4v2 (v = peak velocity of tricuspid regurgitation, m/second), and RAP was empirically estimated to be 10 mmHg [20]. Mean PAP was calculated from the equation: mean PAP = 0.61 ├Ś systolic PAP + 2 mmHg [21].
Right heart catheterization
We routinely performed RHC in our medical intensive care unit using a Swan-Ganz catheter (Swan-Ganz continuous CO/end diastolic volume thermodilution catheter models 777F8 or 777HF8, Edwards Lifesciences, Irvine, CA, USA) and a vigilance monitor (Edwards Lifesciences). We checked hemodynamic variables, such as systolic PAP, mean PAP, diastolic PAP, pulmonary capillary wedge pressure (PCWP), and CO, using the thermodilution method, as well as the response to the vasoreactivity test. PVR was estimated using RHC data and the formula: (mean PAP ŌĆō PCWP) / CO. The cardiac index (CI) was calculated from the following formula: CI = CO / body surface area.
Chest computed tomography
We reviewed chest CT images using medical analysis software (PetaVision). We calculated the pulmonary artery to aorta ratio by measuring the diameter of the main pulmonary artery and the aorta at the level of the bifurcation of the left and right main pulmonary arteries. We repeated this process at least twice to increase accuracy.
Statistical analysis
Data are expressed as median (interquartile range) or as number (%). The clinical characteristics of the patients were analyzed using the Mann-Whitney U test and the chi-square test. Patient survival was analyzed by Cox-regression analysis and the Kaplan-Meier method. A p < 0.05 was significant. All analyses were performed with SPSS version 19 (IBM Co., Armonk, NY, USA).
RESULTS
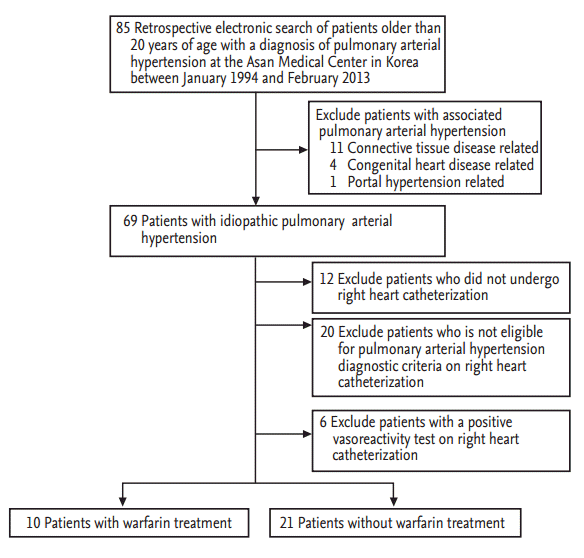
A total of 85 patients with PAH were enrolled initially in our study. We isolated the patients with IPAH by excluding 16 patients with associated PAH (connective tissue disease [11], congenital heart disease [4], and portal hypertension [1]). Among the remaining 69 patients with IPAH, we further excluded 12 patients who did not undergo RHC, 20 who were not compatible with the PAH diagnostic criteria, and six with positive vasoreactivity on RHC. Of the final group of 31 patients with IPAH and a negative vasoreactivity test on RHC, 10 received warfarin (warfarin group) and 21 were treated without warfarin (non-warfarin group) within the first year after the diagnosis (Fig. 1).
The warfarin treatment international normalized ratio target was 1.5 to 2.5. The baseline characteristics of the study patients and both subgroups are presented in Table 1. Our study patients were young and predominantly female. The median time (interquartile range) from onset of symptoms to diagnosis was 19.0 months (interquartile range, 6.0 to 36.0). All PAH treatment drugs other than warfarin that were used within the first year from the IPAH diagnosis are listed in Table 2. Most of the patients received other treatments for IPAH, including conventional therapies (oxygen supplementation, digoxin, and diuretic therapy) and targeted therapies (prostanoids, endothelin receptor antagonists, and phosphodiesterase type 5 inhibitors) [22]. No differences were detected in the baseline characteristics or treatments between the warfarin and non-warfarin groups. The echocardiographic and RHC findings of the patients were compatible with PAH (Table 3). The hemodynamics findings, as measured by RHC, were similar with those of echocardiography. The diameter of the main pulmonary artery was larger than that of the aorta, as shown in Table 4 (median ratio of the pulmonary artery to the aorta, 1.2).
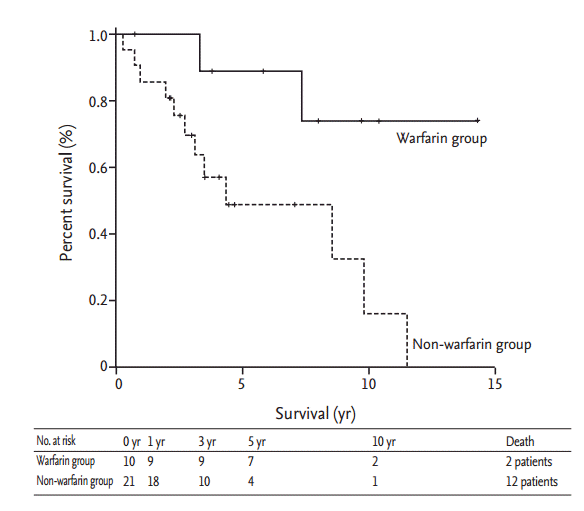
We analyzed survival in our patients using the Kaplan-Meier method (Fig. 2). The median follow-up period was 8.5 years, during which 14 patients died. The overall survival rates of the patients at 1, 3, 5, and 10 years were 90.2%, 79.5%, 62.7%, and 34.8%, respectively. The warfarin group had 2-fold higher mean survival (12.0 years in the warfarin group vs. 6.1 years in the non-warfarin group, p = 0.023). Survival rates at 1, 3, 5, and 10 years were 100.0%, 100.0%, 88.9%, and 74.1%, respectively, in the warfarin group, and 85.7%, 69.7%, 48.9%, and 16.3%, respectively, in the non-warfarin group. We also analyzed factors influencing survival using Cox regression analysis (Table 5). Both univariate and multivariate Cox regression analyses demonstrated that high mean PAP was negatively associated with survival, and use of warfarin was positively associated with survival.
DISCUSSION
This is the first Korean study to evaluate the efficacy of warfarin on long-term survival of patients with IPAH. We analyzed patients with IPAH > 20 years old with negative vasoreactivity confirmed by RHC between January 1994 and February 2013. Because a positive vasoreactivity test is associated with a good prognosis [6] and all IPAH patients with a positive vasoreactivity test were assigned to the non-warfarin group, we excluded such cases and analyzed the survival effect of warfarin in Korean patients with IPAH. As shown in Table 1, the baseline characteristics of the study patients were similar to those from the National Institutes of Health (NIH) and Chinese registries [23,24]. Our study patients were young and predominantly female, and dyspnea was the most common presenting symptom. The NIH registry, the Chinese registry, and our study patients were diagnosed approximately 2 years after symptom onset, showing that an early IPAH diagnosis remains difficult. In our current study, bosentan was not combined with the warfarin treatment (Table 2), as it can significantly reduce maximum prothrombin time (23% to 38%) [25].
We observed that survival in the warfarin group was greater than that in the non-warfarin group. The main pathophysiological findings in patients with IPAH are prothrombotic abnormalities and in situ thrombosis [10]. We believe that warfarin prevents this coagulation cascade from progressing and improves survival of patients with IPAH. No significant differences were observed between the two groups in baseline characteristics, drug treatment, hemodynamic variables measured by echocardiography and RHC, or vessel diameter on chest CT, consistent with previous studies [5-9]. We speculate that the lower WHO functional class, which has a tendency for more use of targeted agents, including sildenafil, iloprost, and ambrisentan in the warfarin group than those in the non-warfarin group, may have affected the survival outcomes of the groups.
As mentioned previously, the survival benefits of anticoagulation therapy in patients with IPAH was recently studied in a large prospective cohort [10]. Although the primary outcomes were similar between this previous study and our current analysis, the patients in that study had different baseline characteristics from those in our series (older, fewer females, higher WHO functional class, and shorter 6-minutes walking distance but lower mean PAP) and were also different from those recorded in most other PAH registries [23,24,26-28]. No prospective randomized controlled studies have been conducted on warfarin as a treatment for PAH; thus, this needs to be addressed in the future.
Here, we evaluated the survival benefits of warfarin in a group of carefully selected patients with IPAH confirmed by RHC with a negative vasoreactivity test, but our study had several limitations. First, our study population may have been subject to selection bias, as it originated from a single tertiary hospital in Korea. Therefore, our results may not be fully applicable to other community-based hospitals. Second, due to the retrospective nature of our analysis, some of the patient data including the RHC and/or chest CT findings were incomplete or difficult to decipher. Third, there may have been a lead-time bias between our two patient groups because of the long study period. Fourth, our study investigated a small sample size, i.e., only 10 warfarin patients and 21 non-warfarin patients. A larger sample size is required to make a more definitive conclusion about the efficacy of warfarin. Finally, our patients with IPAH were diagnosed by a single pulmonologist, which may have underestimated the prevalence of PAH in the initial cohort from which we selected our patients.
However, despite these limitations, this study is based on relatively long-term follow-up data and is the first to evaluate warfarin treatment in Korean patients with IPAH. Our findings suggest that warfarin has significant long-term survival benefits for patients with IPAH. The efficacy of anticoagulation therapy for treating IPAH is somewhat uncertain, but future prospective randomized studies are warranted to address this issue in Korean IPAH cases and patients with this condition in other countries.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





