


To the Editor,
In autoimmune disease, otological symptoms are not unusual. Sensorineural hearing loss (SNHL) is defined as hearing loss of at least 30 dB, in three sequential frequencies within the pure tone audiogram, caused by impairments in the vestibulocochlear nerve (cranial nerve VIII), inner ear, or central processing centers of the brain [1]. SNHL is commonly reported in granulomatosis polyangiitis (GPA; WegenerŌĆÖs granulomatosis), polyarteritis nodosa, Behcet syndrome, rheumatoid arthritis, and systemic lupus erythematosus. SNHL is observed in approximately 8% of GPA cases, but in primary Sj├ČgrenŌĆÖs syndrome (pSS) hearing loss as the primary manifestation is not well known and there are only limited reports [2,3]. Tumiati et al. [4] reported that 46% (14/30) had SNHL in patients with pSS. Ziavra et al. [5] founded SNHL in 22.5% (9/40) of pSS patients. But these reports do not stated about treatment result of patients. We reported that SNHL was first leading symptom that diagnosed with pSS and we successfully treated with high dose corticosteroid.
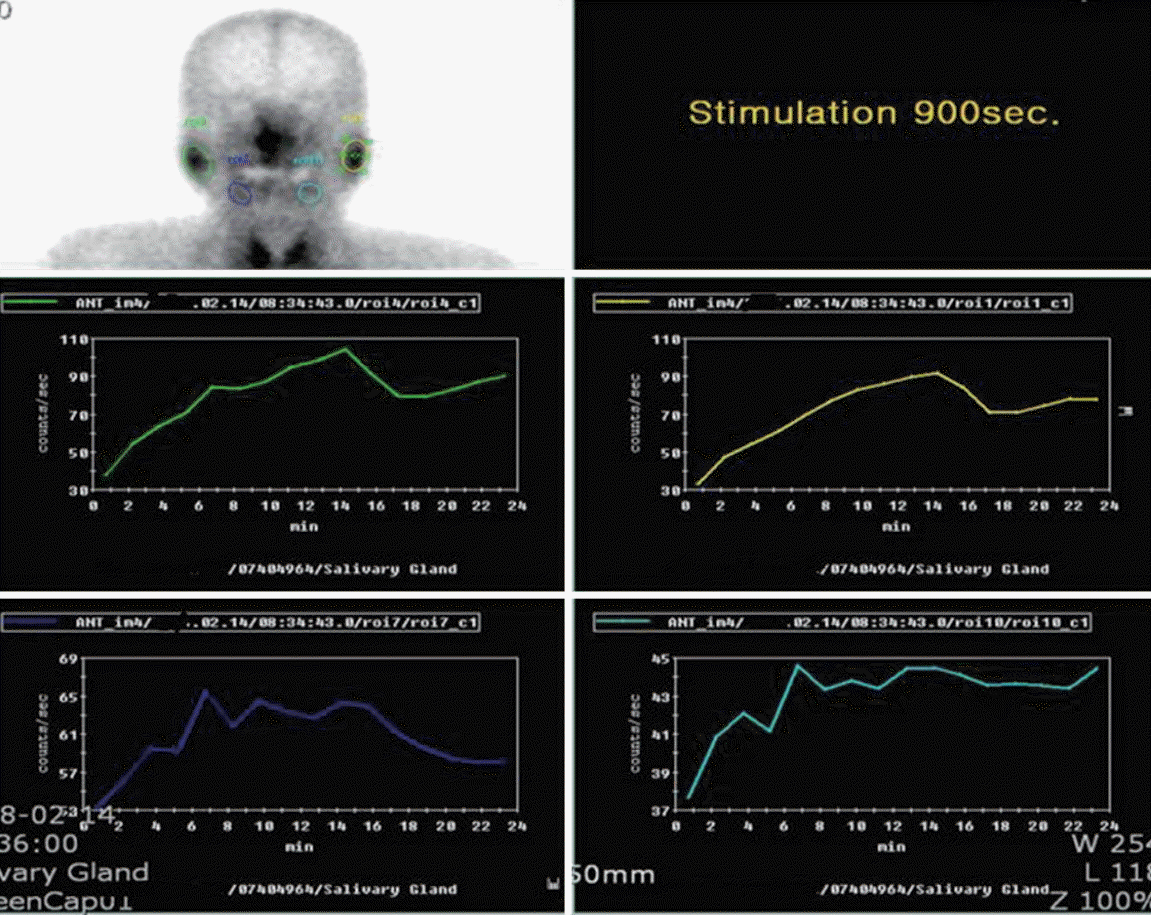
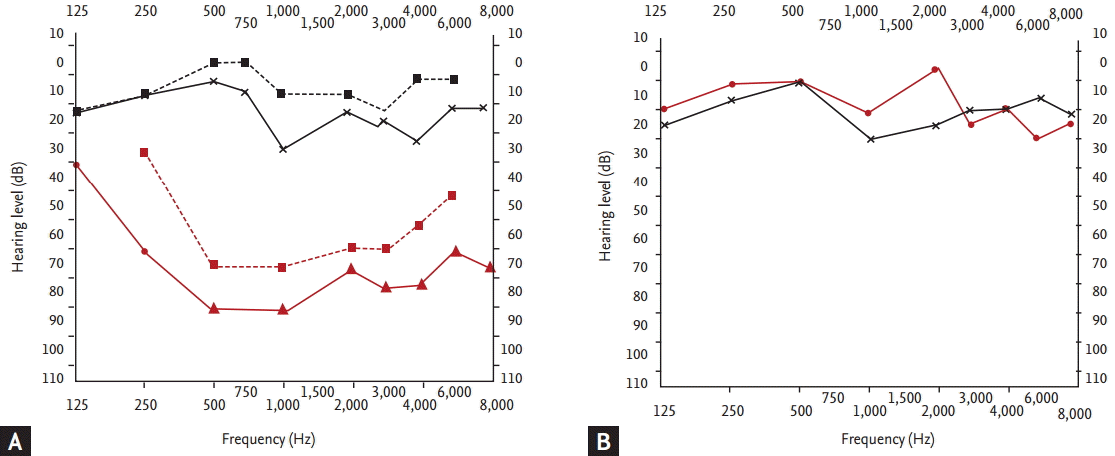
A 62-year-old female was referred with dizziness and sudden-onset hearing difficulties. The patient had been suffering from dry eyes, a dry mouth, and arthritis for several years. There was no history of trauma or otitis. On admission, vital signs including body temperature were within the normal range. Physical examination revealed a regular heart beat without murmur, clear breath sounds without rales, and an absence of pale conjunctivae, pretibial pitting edema, and abdominal and rebound tenderness. Laboratory tests indicated a white blood cell count of 3,980/┬ĄL, with hemoglobin at 12.4 g/dL, and a platelet count of 207,000/mm3. The patientŌĆÖs erythrocyte sedimentation rate was elevated to 53 mm/hr (normal range, 0 to 30), with C-reactive protein at 0.23 mg/dL. Antinuclear antibody was positive, with a speckled pattern and a titer of 1:800. Anti-Ro antibody was 3+; anti-dsDNA antibody, anti-La, anti-cardiolipin, and anti-phospholipid antibodies were all negative. C3 and C4 were in normal range. The serum IgG level was elevated to 2,159 mg/dL (normal range, 870 to 1,700). She had a positive Shirmer test (3 mm after 5 minutes) and Rose Bengal staining. A salivary gland scan with Tc-99m pertechnetate revealed decreased uptake in both submandibular glands, with poor excretion after stimulation, suggesting inflammatory changes (Fig. 1). These findings were compatible with pSS. Upon admission, pure tone audiometry indicated hearing impairment between 500 to 4,000 Hz in the right ear (Fig. 2A). After diagnosis of pSS with SNHL, treatment commenced with high-dose of the methylprednisolone (250 mg) infusion for 5 days; the dose was tapered gradually. After 3 months, pure tone audiometry indicated that hearing in the right ear was in the normal range (Fig. 2B). Sicca symptoms were also improved with supportive care. The patient was maintained on a low-dose steroid (prednisolone 7.5 mg) and hydroxychloroquine (300 mg).
Although viral, vascular, and immunologic theories have all been proposed in the pathogenesis of SNHL, the immunologic theory has more recently been considered as the most common cause. This theory is based on antibody activity and cytotoxic T-cell-mediated apoptosis in the inner ear [1]. Several studies have reported that anti-phospholipid antibody and anti-heat shock protein-70 is associated with SNHL. It has been suggested that these autoantibodies induce thrombosis in the labyrinthine vessels, thereby causing damage to the inner ear resulting in SNHL [1].
pSS is a chronic autoimmune disease characterized by lymphocyte infiltration of both the salivary and lacrimal glands, resulting in dry eyes and a dry mouth. Otologic symptoms may be an early indicator of pSS, but are also a less-common feature of other autoimmune disorders [3]. Although pSS patients tend to have a higher prevalence of sensorineural hearing impairment compared with the general population, no evidence of damage to the central auditory pathways was observed in our patient [4]. The majority of pSS patients exhibit hearing impairments of cochlear origin, principally at high frequencies. Autoantibodies of both cardiolipin and M3 muscarinic receptors in the sera are suspected to play a pathogenetic role in the progressive hearing loss of pSS patients [3]. Sensorineural damage may be attributable to vasculitis or neuritis, or may represent an ototoxic effect of the drugs used to treat pSS [4]. Ziavra et al. [5] reported a correlation between SNHL and pSS duration, but no correlation between SNHL and age, systemic manifestations or the presence of autoantibodies. Increased expression of anti-cardiolipin, anti-endothelial cells, and anti-neutrophil cytoplasmic antibodies may correlate with clinical manifestations, including vasculitis, glomerulonephritis and hearing loss [3]. Tumiati et al. [4] reported a higher prevalence of anticardiolipin antibodies in pSS patients with SNHL. In our patient, there was no history of medication use that could have affected the otologic system; anti-cardiolipin and anti-phospholipid antibodies were also all negative.
Published evidence supports the hypothesis that immunologic impairment is the most common cause of SNHL. Therefore, corticosteroids or immunosuppressants are recommended for SNHL in autoimmune disease [1]. In a multi-center randomized controlled trial, methotrexate was no more effective than placebo in patients with progressive bilateral hearing loss. Corticosteroids and nonsteroidal anti-inflammatory drugs are usually effective treatments of SNHL and systemic complications [3]. Several case reports have demonstrated the benefits of immunosuppressants (e.g., cyclophosphamide, azathioprine, and mycophenolate mofetil) for patients with severe and progressive SNHL [1]. Treatment with a chimeric, monoclonal anti-CD20 antibody (rituximab) inhibits B-cell proliferation, suggesting good treatment potential [3]. Emerging reports on the efficacy of tumor necrosis factor-╬▒ blockade in SNHL patients emphasize the role of cellular immunity [2]. Although SNHL was characterized by a high spontaneous recovery rate in a pilot study, idiopathic hearing loss may represent the initial manifestation of systemic vasculitis, including pSS [1]. Therefore, early referral to a rheumatologist should be considered for prompt diagnosis of underlying autoimmune disease and subsequent initiation of therapy [2].
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





