


INTRODUCTION
Asthma is characterized by inflammation of the airways, reversible airflow obstruction, and airway hyper-responsiveness. Airway remodeling can be defined as changes in the composition and organization of the cells and extracellular constituents of the airway wall [1]. These structural changes increase airway wall thickness, resulting in a decline in lung function and resistance to asthma treatment [2]. Airway remodeling is characterized by subepithelial fibrosis, increased airway smooth muscle mass, goblet cell hyperplasia, and angiogenesis [3].
Inhaled corticosteroids remain the mainstay of anti-inflammatory treatment in asthma. Many studies have investigated the effects of inhaled corticosteroids on airway remodeling. Chetta et al. [4] reported that high doses of fluticasone affected airway remodeling by reducing both submucosal vascularity and basement membrane thickness. It has been demonstrated that inhaled corticosteroids prevent airway remodeling through inhibition of thickening of the airway smooth muscle layer [5-7]. However, the effect of inhaled corticosteroids on subepithelial fibrosis is controversial [8,9].
We previously reported that a tyrosine kinase inhibitor can ameliorate both airway inflammation and airway remodeling in a chronic asthma model [10,11]. Nilotinib (Tasigna, Novartis Pharmaceuticals, Basel, Switzerland) is a small-molecule tyrosine kinase inhibitor developed to treat chronic myeloid leukemia [12]. Nilotinib inhibits the tyrosine kinase activity of not only the Abl kinases but also c-kit and the platelet-derived growth factor (PDGF) receptor [12]. Additionally, nilotinib targets transforming growth factor ╬▓ (TGF-╬▓), a profibrotic cytokine thought to play important roles in fibrotic tissue remodeling in asthma, and has been investigated in many fibrotic diseases [13-16].
The aim of this study was to compare the effects of fluticasone and nilotinib on airway remodeling in a chronic asthma model. We also examined whether co-treatment with nilotinib and fluticasone had any synergistic effect in the prevention of airway remodeling.
METHODS
Sensitization and antigen challenge protocol
Female 6-week-old BALB/c mice weighing 20 to 25 g were used (n = 7 to 9 mice per group; Orient Bio Experimental Animal Center, Seongnam, Korea). Mice were assigned to one of the following five treatment groups: (1) control; (2) ovalbumin (OVA, grade V; Sigma-Aldrich, St. Louis, MO, USA) challenge; (3) OVA challenge plus 100 ╬╝g/kg fluticasone; (4) OVA challenge plus 80 mg/kg nilotinib; or (5) OVA challenge plus 100 ╬╝g/kg fluticasone and 80 mg/kg nilotinib. Mice were immunized by subcutaneous injection on days 0, 7, 14, and 21 with 25 ╬╝g of OVA absorbed to 1 mg of aluminum hydroxide (Sigma-Aldrich, Milwaukee, WI, USA) in 200 ╬╝L of phosphate-buffered saline (PBS). Intranasal OVA challenges (20 ╬╝g/50 ╬╝L in PBS) were administered on days 27, 29, and 31 under isoflurane (Vedco, St. Joseph, MO, USA) anesthesia. Intranasal OVA challenges were then repeated twice per week for 3 months. Allergen-sensitized mice were treated with fluticasone by intranasal administration and/or nilotinib by oral gavage. Fluticasone and/or nilotinib were given five times per week starting on day 35 and then during the OVA challenges for 3 months. Mice were sacrificed 24 hours after the final OVA challenge, and lung tissues were analyzed. All animal experimental protocols were approved by The Catholic University of Korea, Animal Subjects Committee.
Bronchoalveolar lavage
Mice were sacrificed by CO2 asphyxiation. The trachea was exposed and cannulated with silicone tubing attached to a 23-gauge needle on an 800-╬╝L tuberculin syringe. After instillation of 1 mL sterile PBS through the trachea into the lung, BAL fluid was withdrawn. The total number of cells in the BAL fluid was counted using a hemocytometer. The BAL fluid was cytospun (7 minutes, 2,000 rpm) onto microscope slides and stained with Diff-Quick (Sysmax, Kobe, Japan). The percentages of macrophages, eosinophils, lymphocytes, and neutrophils in the BAL fluid were obtained by counting 400 leukocytes on randomly selected portions of the slides using light microscopy. Supernatants were stored at ŌĆō70┬░C until the day of measurement.
Enzyme-linked immunosorbent assay
The concentrations of interleukin 4 (IL-4), IL-5, IL-13, and TGF-╬▓1 were measured in the BAL fluid with enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA). The acid-activated latent form of TGF-╬▓1 was measured. The protocol followed was according to the manufacturersŌĆÖ instructions. The sensitivity of the ELISA was as follows: (1) IL-4, 2 pg/mL; (2) IL-5, 7 pg/mL; (3) IL-13, 1.5 pg/mL; and (4) TGF-╬▓1, 15.6 pg/mL.
Immunohistochemistry
Six-micron-thick sections of lung from each paraffin block were deparaffinized with xylene and rehydrated in an ethanol series. For immunohistochemical detection of ╬▒-smooth muscle actin, the lung sections were incubated overnight at 4┬░C with a primary monoclonal antibody against ╬▒-smooth muscle actin (Sigma-Aldrich) or mouse serum as a negative control instead of the primary antibody. Immunoreactivity was detected by sequential incubations of lung sections with a biotinylated secondary antibody, followed by peroxidase reagent and the 3-amino-9-ethylcarbazole (AEC) chromogen. The immunostaining area of ╬▒-smooth muscle actin in each paraffin wax-embedded lung was outlined and quantified using a light microscope attached to an image analysis system (BX50, Olympus, Tokyo, Japan). Results are expressed as the area of immunostaining per micron length of basement membrane of bronchioles (internal diameter, 150 to 200 ╬╝m). At least 10 bronchioles were counted on each slide.
Hydroxyproline analysis
Aliquots of lung tissue (60 mg) from each mouse were used for the hydroxyproline assays. A sample of lung homogenate was added to 250 ╬╝L of 12 N HCl for 16 hours at 110┬░C. Following centrifugation, 25 ╬╝L of each supernatant was assayed. A total of 25 ╬╝L of citrate/acetate buffer (5% citric acid, 7.2% sodium acetate, 3.4% sodium hydroxide, and 1.2% glacial acetic acid) and 500 ╬╝L of chloramine-T solution (1.41 g chloramine-T, 26 mL n-propanol, 20.7 mL distilled water, and 53.3 mL citrate/acetate buffer) were added to a 25 ╬╝L sample of the digested lung. The resulting samples were then incubated at room temperature for 20 minutes before 500 ╬╝L of EhrlichŌĆÖs solution (4.5 g Žü-dimethylaminobenzaldehyde, 18.6 mL n-propanol, and 7.8 mL 70% perchloric acid) was added. These samples were incubated for 15 minutes at 65┬░C, and cooled samples were read at 550 nm in a spectrophotometer. Hydroxyproline concentrations were calculated from a standard curve.
Measurement of smooth muscle area
The immunostained area of ╬▒-smooth muscle actin in each paraffin wax-embedded lung was outlined and quantified using a light microscope attached to an image analysis system (BX50, Olympus). Results are expressed as the area immunostained per micrometer length of basement membrane of bronchioles (internal diameter, 650 to 750 ╬╝m). At least 10 bronchioles were counted on each slide.
Proliferation assay
Cell proliferation was quantified by the colorimetric XTT-based assay kit (Boehringer Mannheim, Mannheim, Germany). A lung fibroblast cell line (HFL-1) was obtained from the American Type Culture Collection (No. CCL-153, Rockville, MD, USA). Cells were seeded in 96-well microtiter plates (Greiner, Frickenhausen, Germany) at a concentration of 5 ├Ś 103 cells/mL. Cells were incubated in DulbeccoŌĆÖs modified EagleŌĆÖs medium containing 10% fetal bovine serum and 1% penicillin-streptomycin at 37┬░C in a humidified atmosphere of 5% CO2 for 24 hours, then serum-starved for 24 hours. Nilotinib or fluticasone was co-administered with PDGF-AA or stem cell factor (SCF), and cells were incubated for 72 hours. Next, 50 ╬╝L of XTT solution was added for 4 hours at 37┬░C. Absorbance was measured at 450 nm in a Dynatech MR 3.13 MicroELISA reader (Dynex Technologies, Ashford, UK).
RESULTS
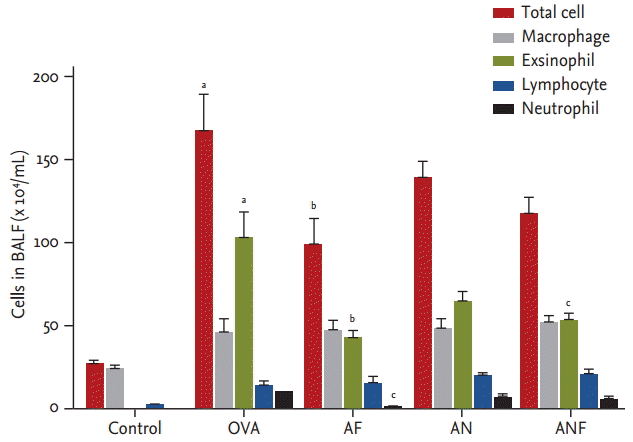
Effects of fluticasone and nilotinib on airway inflammation
Repeated OVA challenge induced a significant increase in the number of total cells and eosinophils in the BAL fluid. Fluticasone treatment in OVA-challenged mice reduced significantly the numbers of total cells, eosinophils, and neutrophils in BAL fluid (Fig. 1). There was no significant difference in the numbers of total cells, macrophages, eosinophils, lymphocytes, or neutrophils between the OVA and nilotinib group. The combination of fluticasone and nilotinib treatment reduced only the number of eosinophils.
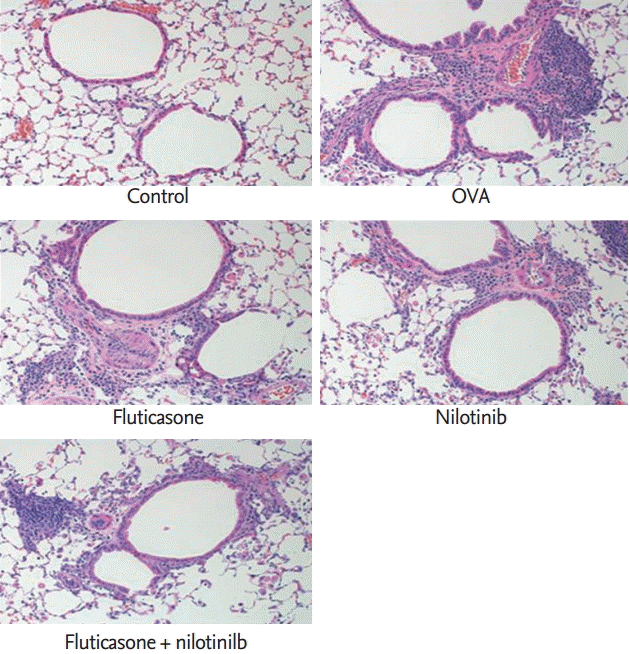
Effects of fluticasone and nilotinib on lung histopathology
Histological lung sections demonstrated that repeated OVA challenge induced marked increases in subepithelial, peribronchial, and perivascular inflammation compared with the control group. In sensitized mice, the airway architecture was distorted by epithelial folding, subepithelial fibrosis, and lumen narrowing compared with control mice. These histopathological inflammatory changes were reduced after treatment with fluticasone and nilotinib, and co-administration of both drugs showed synergistic effects (Fig. 2).
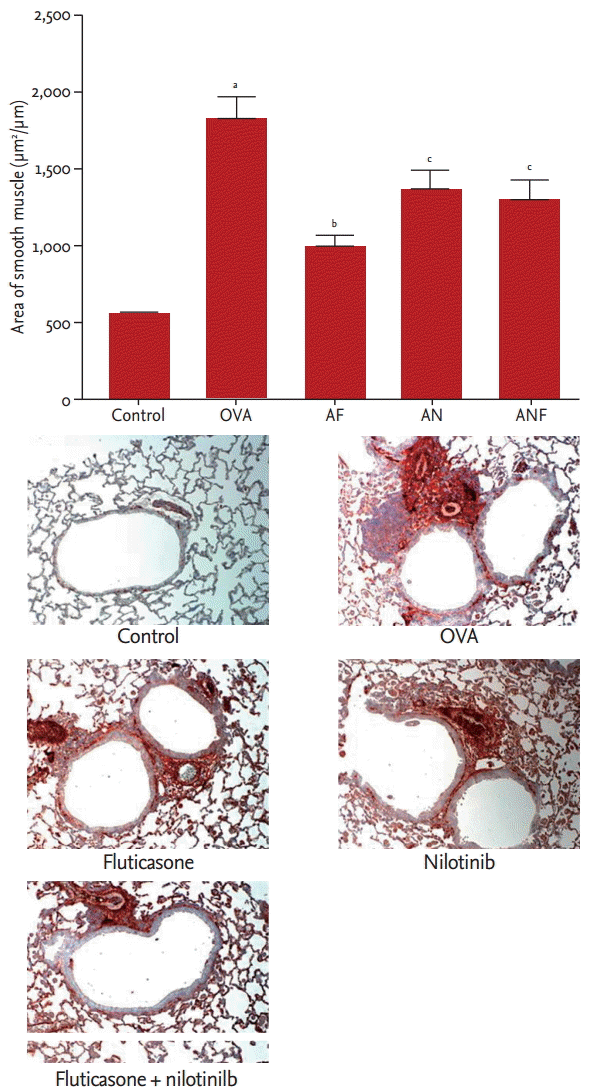
Effects of fluticasone and nilotinib on the area of the airway smooth muscle layer
Repeated OVA challenge resulted in a significant increase in the area of peribronchial ╬▒-smooth muscle actin immunostaining (control, 0.57 ┬▒ 0.06 ╬╝m2/╬╝m vs. OVA, 1.80 ┬▒ 0.67 ╬╝m2/╬╝m; circumference of bronchiole, respectively; p < 0.01). Fluticasone significantly reduced the area of peribronchial ╬▒-smooth muscle actin staining in mice subjected to repeated OVA challenge (fluticasone, 0.10 ┬▒ 0.30 vs. OVA, 1.80 ┬▒ 0.67, respectively; p < 0.01). Nilotinib and the combination of nilotinib and fluticasone also significantly reduced the area of peribronchial ╬▒-smooth muscle actin staining in repeatedly OVA-challenged mice (nilotinib, 1.35 ┬▒ 0.59 vs. OVA, 1.80 ┬▒ 0.67, respectively; nilotinib + fluticasone, 1.29 ┬▒ 0.53 vs. OVA, 1.80 ┬▒ 0.67, respectively; p < 0.05 for both) (Fig. 3).
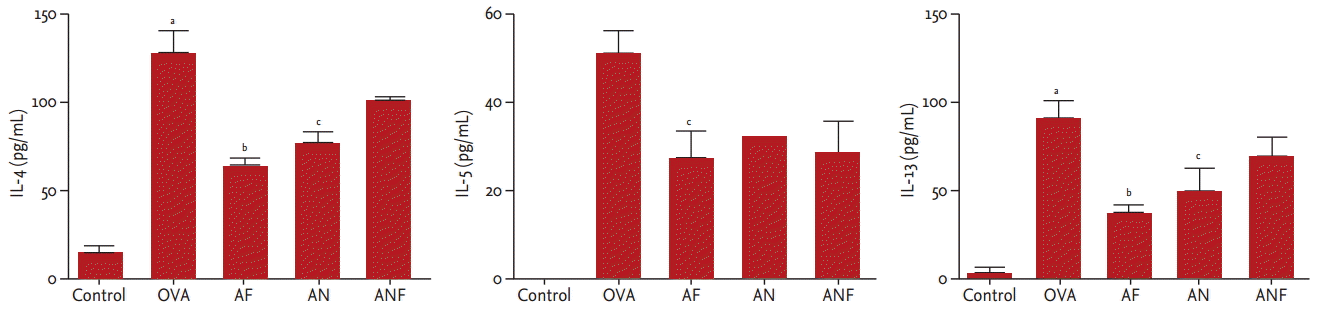
Effects of fluticasone and nilotinib on IL-4, IL -5, and IL -13 levels
Repeated OVA challenges induced significant increases in the levels of IL-4, IL-5, and IL-13 in BAL fluid. Fluticasone treatment significantly reduced the levels of IL-4 (119.34 ┬▒ 51.87 vs. 60.31 ┬▒ 14.31, p < 0.01), IL-5 (49.34 ┬▒ 16.79 vs. 26.49 ┬▒ 19.35, p < 0.05), and IL-13 (88.37 ┬▒ 33.54 vs. 37.07 ┬▒ 17.25, p < 0.01). Nilotinib treatment significantly reduced the levels of IL-4 (119.34 ┬▒ 51.87 vs. 72.76 ┬▒ 18.69, p < 0.05) and IL-13 (88.37 ┬▒ 33.54 vs. 49.20 ┬▒ 33.80, p < 0.05). Co-treatment with nilotinib and fluticasone significantly reduced the IL-5 level (49.34 ┬▒ 16.79 vs. 30.99 ┬▒ 18.89, p < 0.05) (Fig. 4).
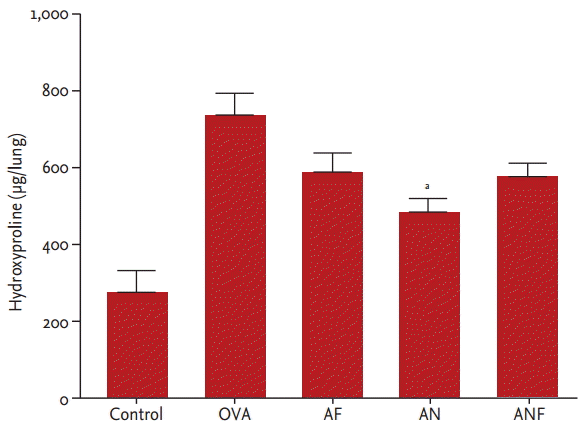
Effects of fluticasone and nilotinib on collagen levels
Collagen deposition was assessed by measuring the hydroxyproline content in lung tissues. The level of hydroxyproline was measured in ╬╝g/lung. Repeated OVA challenges induced a significant increase in the level of hydroxyproline (control, 2.75 ┬▒ 1.54 vs. OVA, 7.16 ┬▒ 2.14, respectively; p < 0.01). Nilotinib treatment significantly reduced the level of hydroxyproline (nilotinib, 4.78 ┬▒ 1.13 vs. OVA, 7.16 ┬▒ 2.14, respectively; p < 0.050) (Fig. 5). The level of hydroxyproline showed a trend towards reduction with fluticasone and combined fluticasone and nilotinib treatment.
Effects of fluticasone and nilotinib on TGF-╬▓1 levels
Repeated OVA challenges induced a significant increase in the level of TGF-╬▓1 (control, 659.46 ┬▒ 276.84 vs. OVA, 1661.34 ┬▒ 408.58, respectively; p < 0.01). Nilotinib treatment significantly reduced the level of TGF-╬▓1 (nilotinib, 1,070.66 ┬▒ 239.58 vs. OVA, 1,661.34 ┬▒ 408.58, respectively; p < 0.01) (Fig. 6). However, neither fluticasone alone nor co-administration with nilotinib reduced the level of TGF-╬▓1.
Effects of fluticasone and nilotinib on growth of lung fibroblasts stimulated by PDGF and SCF
Nilotinib and the combination of nilotinib and fluticasone inhibited PDGF-AA-stimulated cell proliferation. However, fluticasone alone had no inhibitory effect on PDGF-AA-stimulated cell proliferation. The addition of fluticasone and/or nilotinib significantly inhibited SCF-induced cell proliferation (Fig. 7).
DISCUSSION
In this study, we demonstrated that both fluticasone and nilotinib treatment can attenuate airway smooth muscle thickening. However, only nilotinib suppressed fibrotic changes, demonstrating inhibition of collagen deposition as measured by hydroxyproline analysis. Pro-inflammatory cells, such as eosinophils, and several cytokines, such as IL-4, IL-5, and IL-13, induced by repeated OVA challenge, were reduced by fluticasone. We confirmed that fluticasone had anti-inflammatory effects while nilotinib was more effective in reducing the levels of TGF-╬▓1. Thus, we suggest that fluticasone and nilotinib have therapeutic effects on airway smooth muscle remodeling in different ways.
Inhaled corticosteroids are the mainstay of anti-inflammatory treatment in asthma, but their effect on airway fibrosis is debatable [8,9]. This may be because airway remodeling in asthma is a consequence of mechanical stimuli as well as inflammation. The response of airway epithelial cells to acute compressive stress during bronchoconstriction results in the release of growth factors, such as early growth response-1 and TGF-╬▓1 [17-21]. Our results support these findings in that inhaled corticosteroids did not affect collagen deposition or the level of TGF-╬▓1. Thus, novel therapeutic agents targeting airway fibrosis may offer added benefit to inhaled corticosteroids for preventing airway remodeling.
SCF, the ligand for the c-kit receptor, is expressed by inflammatory cells in the airways [22]. SCF is an important growth factor for mast cells, so binding of SCF and c-kit activates dysregulated mast cell-related disorders, including asthma. Thus, tyrosine kinase inhibitors targeting c-kit activity have been investigated in asthma [23]. PDGF is a chemoattractant for the migration of human airway smooth muscle cells, contributing to airway remodeling in asthma [24]. Tyrosine kinase inhibitors block the tyrosine kinase activity of PDGF receptors and thereby can prevent PDGF-induced PDGFR autophosphorylation and signaling [13]. Berlin et al. [25] showed that imatinib, the most widely recognized tyrosine kinase inhibitor, has a significant effect on chronic peribronchial allergen-induced fibrotic remodeling. Additionally, tyrosine kinase inhibitors simultaneously target two major profibrotic pathways, TGF-╬▓ and PDGF signaling [26]. We previously showed that c-kit and PDGFR tyrosine kinase inhibitors such as imatinib and nilotinib attenuated airway remodeling in a chronic asthma model [10,11]. In the present study, nilotinib treatment significantly decreased collagen deposition. To investigate the inhibitory effect of nilotinib treatment on fibrotic remodeling, we assessed the growth of lung fibroblasts stimulated by PDGF and SCF in vitro. Treatment with nilotinib inhibited this cell proliferation significantly. We confirmed that the anti-fibrotic effect of nilotinib was mediated by inhibition of the c-kit and PDGFR pathways.
TGF-╬▓ is a profibrotic cytokine, and TGF-╬▓ isoforms have several roles in the regulation of airway inflammation and the remodeling process in asthma [27]. The levels of TGF-╬▓ are increased in the airways in asthma and elevated further in response to allergen exposure [28]. TGF-╬▓ stimulates the differentiation of fibroblasts to myofibroblast cells and their proliferation, resulting in extracellular matrix proteins [29,30]. TGF-╬▓ also enhances airway smooth muscle proliferation, mediated by extracellular matrix-integrin interactions [31,32]. Thus, modulation of TGF-╬▓ represents a potential therapeutic target for the prevention or reversal of airway remodeling [33]. Some studies have shown that nilotinib decreases the level of TGF-╬▓. Liu et al. [34] suggested that nilotinib treatment inhibited not only PDGF and Abl signaling but also TGF-╬▓ cytokines in vitro. In a murine lung injury model, TGF-╬▓ expression was decreased by nilotinib treatment [15]. The results of the present study correspond well with these results. Nilotinib effectively attenuated airway smooth muscle hyperplasia and collagen deposition, decreasing levels of TGF-╬▓1.
Currently, treatment of asthma is focused on amelioration of inflammation and the reversal of bronchoconstriction using inhaled corticosteroids. However, treating only the inflammatory component using inhaled corticosteroids without controlling airway caliber does not prevent airway remodeling, resulting in the loss of lung function [35]. Although several clinical studies suggest that inhaled corticosteroids can inhibit airway remodeling, their effectiveness in reversing established remodeling is limited [7,35,36]. Furthermore, there is no effective therapeutic agent known to prevent or reverse airway remodeling, although the reversibility of airway remodeling has been suggested by studies conducted in animal models [37]. Given this, nilotinib may be a promising agent to reverse airway remodeling in asthma. Although our study did not show a synergistic effect of nilotinib and fluticasone in airway remodeling, each agent had therapeutic effects in remodeling through anti-inflammatory and anti-fibrotic pathways, respectively. The combination of nilotinib and inhaled corticosteroids may be a therapeutic option to reverse airway remodeling in asthma patients. Further study is needed to determine any synergistic effect of these agents, and a larger-scale clinical trial is required for asthma patients resistant to steroid treatment.
In conclusion, we demonstrated that fluticasone and nilotinib treatment inhibited smooth muscle thickening in relation to airway remodeling in a chronic asthma model. Only nilotinib reduced subepithelial fibrosis, through inhibition of PDGFR and c-kit. Our results provide evidence for the therapeutic potential of nilotinib in the treatment of chronic asthma resistant to steroid treatment. Further studies regarding the effect of nilotinib on airway remodeling are needed.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





