


INTRODUCTION
Ischemia is a common cause of ARF in hospitalized patients. Ischemic ARF is a life threatening illness that continues to have a high mortality rate of 50~80% for the patients in intensive care settings1). A better understanding of ischemic ARF may result in interventions that can help avoid hemodialysis, shorten the course of ischemic ARF and improve patient survival.
The pathophysiology of ischemic ARF involves inflammation, in addition to the tubular and vascular factors1). Recent studies have indicated the involvement of leukocytes, adhesion molecules, chemokines and cytokines in ischemic ARF2-5). Specific inflammatory cells release cytokine/chemokines that contribute to medullary vascular congestion and the cytotoxic injury to epithelial cells6). Some studies have suggested that neutrophils play an important role in ischemic ARF7-9), while other studies have not10, 11). Although some findings suggest the direct or indirect involvement of T cells in ischemic ARF12-14), other studies have raised doubt about such involvement15). It has recently been suggested that macrophages play a role in ischemic ARF. It has been shown that significant monocyte/macrophage adhesion and infiltration occurs in the outer strip of the outer medulla as early as 24 h post ischemic reperfusion in a rat model16, 17). However, there have been no reports regarding the time-course infiltration of macrophages and its relation to the changes of renal function after ischemic insult.
The present study examined the time-course of macrophage infiltration in a mouse model of ischemic ARF. This study investigated macrophage infiltration and whether the depletion of macrophages affected ischemic ARF.
MATERIALS AND METHODS
Animals
This study used male C57BL/6 mice aged 8-10 weeks (Jackson Laboratories, Bar Harbor, Maine, USA).
Ischemia protocol
Mice weighing 20~25 g each were anesthetized with an intraperitoneal injection of Avertin (2,2,2-tribromoethanol: Sigma-Aldrich, Milwaukee, Wisconsin, USA). A midline incision was made and the renal pedicles were bilaterally clamped for 22 min with using microaneurysm clamps. The clamps were then removed and the kidneys were observed to ensure they had regained their original color, which indicated the restoration of blood flow. The abdomen was then closed in two layers. The ischemia time that was chosen provided a reversible model of ischemic ARF and it didn't cause animal mortality. This method resulted in a serum creatinine peak at 24-48 h after reperfusion commenced, and the serum creatinine levels gradually returned to normal within 5-7 d. The control mice underwent sham surgery that consisted of the same surgical procedure except that clamps were not applied. During the 24-h reperfusion period, the animals were kept in an incubator at 29Ōäā. Blood samples were obtained by cardiac puncture at 2, 8, 16 and 24 h after surgery. The BUN and creatinine levels were measured using an Astra Autoanalyser (Beckman Instruments Inc, Fullerton, California, USA). Each time point measurement represents the mean┬▒the standard error of the mean (SEM) from 6 mice.
Immunofluorescence method
Kidney tissues were embedded in an optimal cutting temperature (O.C.T.); they were snap-frozen in liquid nitrogen and then stored at -80Ōäā until sectioning. The cut cryostat sections (5 ┬Ąm) were fixed in 70% acetone/30% methanol for 5 min, air dried for 5 min, rinsed in phosphate-buffered solution (PBS) for 3 min three times, soaked in 3% paraformaldehyde for 10 min and then rinsed in PBS for 3 min three times. To determine the CD 11b expression, the sections were preincubated with 10% donkey serum for 30 min, and they were next incubated with anti-mouse CD 11b antibody (1:50 dilution in 5% donkey serum in PBS) for 1 h at room temperature; they were rinsed in PBS for 5 min three times and then incubated in anti-donkey horseradish peroxidase (HRP) secondary antibody in 5% donkey serum in PBS. The sections were counterstained by incubating them with 7:1000 Cy3, 1:1,000 wheat germ agglutinin and Alexa Fluor 488 conjugate (green) to identify the cytoplasm and they were incubated with 3:1,000 Hoechst 33,342 (blue) for 60 min at room temperature to identify the nucleus. They were next rinsed in PBS for 5 min three times and then they were rinsed in water for 2 min; they were placed on a slide with mounting medium and a coverslip. The fluorescent images were captured using a digital camera and these were merged with the phase-contrast microscope images. To measure macrophage infiltration, the kidney sections were stained to determine the number of cells expressing CD 11b. Fifteen randomly chosen high-powered fields (├Ś400) of the corticomedullary junction were assessed by a renal pathologist, who was kept "blind: as to how the sections were treated. Tissues from the control (n=4) and ischemia mice (2, 8, 16 and 24 h; n=4 for each time point) were examined.
Preparation of liposomes
Clodronate [dichloromethylene bisphosphonate (Cl2MBP)] was prepared according to the method of van Rooijen et al18). Phosphatidylcholine [86 mg; egg lecithin (20 mg/mL) in chloroform] and cholesterol (8 mg; 8 mg/10 mL chloroform) were evaporated via rotation under reduced pressure (vacuum pump; Rotations evaporator R-114, Buchi Labortechik, Flavil, Switzerland). A suspension of 2.5 g Cl2MBP in 10 mL PBS was stored under N2 for 2 h; this was sonicated and then stored overnight at 4Ōäā. The liposomes were centrifuged for 30 min (10,000 g, 4Ōäā) and they were resuspended in 4 mL PBS before use.
In vivo depletion of macrophages
Clodronate (100 ┬ĄL/10 g mouse) was injected via a tail vein 5 days before ischemia induction and again 2 days before ischemia induction. At 24 h post-ischemic reperfusion, the mice' blood samples were analyzed for monocytes, and the spleens were prepared for flow cytometry as we have previously described15). Empty liposomes, which did not containing clodronate, were prepared in the same manner as the LECs, and the empty liposomes were used as the vehicle (V). Staining was detected by a Beckton-Dickinson FACsCalibur (BD Immunocytometry Systems, San Jose, CA) flow cytometer. CellQuest Software (BD Immunocytometry Systems) was used to analyze the flow cytometry data of the mice that were treated with LECs. Following ischemia induction, the serum creatinine levels were determined.
Statistical analyses
The values are expressed as means┬▒SEMs. The non-normally distributed data was analyzed by the nonparametric unpaired Mann-Whitney test. Multiple-group comparisons were performed by the Kruskal-Wallis test and using post-tests. A p value < 0.05 was considered to indicate a significant difference.
RESULTS
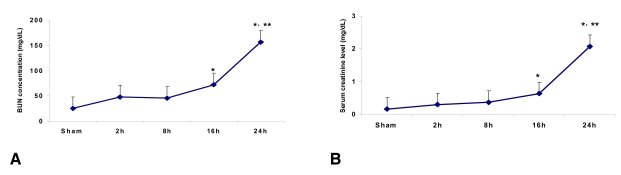
Time course of the serum creatinine and BUN levels following ischemia
The serum creatinine level in the control mice (n=6) was 0.17┬▒0.03 mg/dL at 24 h after sham surgery. The serum creatinine levels at 2, 8, 16 and 24 h from the ischemia mice were 0.30┬▒0.06, 0.37┬▒0.1, 0.63┬▒0.03 (p<0.05 vs. the control and at 2 h and 8 h post-ischemia) and 2.07┬▒0.12 mg/dL (p<0.05 vs. the control and at 2 h, 8 h and 16 h post-ischemia) (n=6 for each time point), respectively. The BUN level in control mice (n=6) was 25.0┬▒2.0 mg/dL at 24 hr after sham surgery. The BUN levels at 2, 8, 16 and 24 h from the ischemia mice were 48.0┬▒2.8, 45.6┬▒4.2, 72.0┬▒3.0 (p<0.05 vs. control and 2 h and 8 h post-ischemia) and 156.0┬▒8.5 mg/dL (p<0.05 vs. the control and at 2 h, 8 h and 16 h post-ischemia) (n=6 for each time point), respectively (Figure 1A and 1B).
Immunofluorescence staining for CD 11b to determine the time-course of macrophage infiltration in the control and ischemic tissues
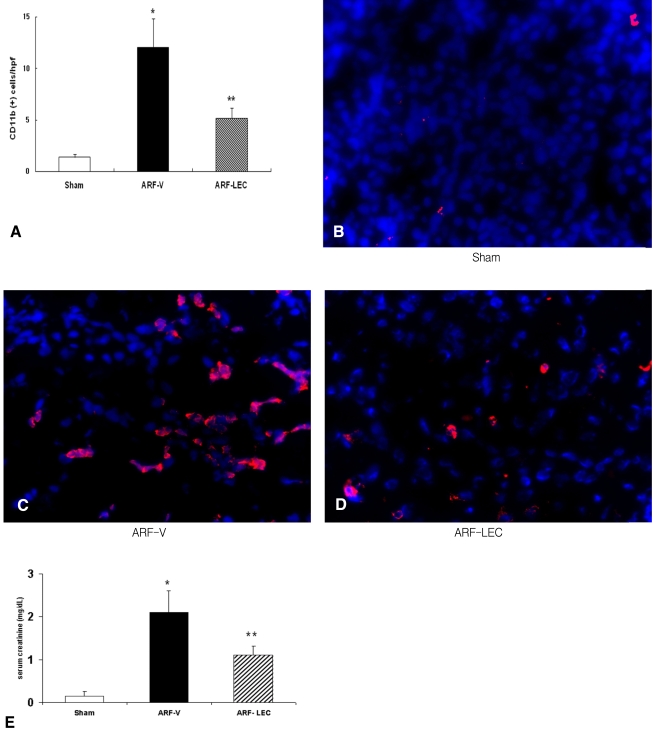
The number of CD 11b-postive cells in the sections from the control and at 2, 8, 16 and 24 h from the ischemia mice was 0.2┬▒0.1, 3.5┬▒0.3, 2.9┬▒0.4, 13.8┬▒1.5 and 11.3┬▒0.7, respectively (n=4 for each time point) (Figure 2A). This data showed there was a significant increase in the number of CD 11b-positive cells in the 2 h ischemia mice compared to the controls (p<0.01), and that macrophage infiltration continued to increase to 16-24 h after ischemia was induced. The representative sections are shown in Figures 2B-F.
Immunofluorescence staining for CD 11b to determine the location of macrophages in the ischemic tissue
We determined the number of CD 11b-positive cells in various kidney regions. This analysis showed there were 1.1┬▒0.3, 13.2┬▒0.9 and 1.6┬▒0.3 CD 11b-positive cells in the 24 hr post-ischemia sections from the cortex, outer medulla and inner medulla, respectively (p<0.01, n=4 for each group, Figure 3A). The representative images are shown in Figures 3B-D.
Renal function after macrophage depletion with using clodronate
The numbers of blood monocytes (106/L) were 55┬▒18 in the sham-operated mice and 145┬▒38 in the ischemic ARF with vehicle (ARF-V) mice and 82┬▒24 in the ischemic ARF with clodronate (ARF-LEC) mice. There was a significant decrease of the blood monocytes in the ARF-LEC mice compared to the ARF-V mice. Administering LECs resulted in a 70% reduction of the macrophages in the spleen (data not shown) and a significant decrease of macrophage infiltration in the kidney of the ischemic ARF mice (*p<0.01 vs. the sham operated, ARF-V mice, **p<0.05 vs. the sham operated mice, n=4 for each group) (Figure 4A). The representative images are shown in Figures 4B-D. The ARF-V mice had higher serum creatinine levels compared to the sham operated mice and the ARF-LEC mice (0.20┬▒0.01 vs 2.10┬▒0.79 vs 1.09┬▒0.67 mg/dL, respectively, *p<0.05, n=8). The ARF-LEC mice had lower serum creatinine levels compared to the ARF-V mice (1.09┬▒0.67 vs. 2.10┬▒0.79 mg/dL, respectively, **p<0.05, n=8 for each group) (Figure 4E).
DISCUSSION
While the previous studies have indicated that macrophages play a role in ischemic ARF, a causal relation has yet to be reported3). Despite that the classical paradigms suggest that macrophages are involved in a destructive proinflammatory response, recent studies have suggested that macrophages may be responsible for restoration of normal function19). A number of studies have demonstrated that monocytes/ macrophages 16, 20, 21), as well as macrophage-associated cytokines such as IL-1, IL-6 and transforming growth factor (TGF)-╬▓21), and monocyte/macrophage chemoattractants such as interferon (IFN) ╬│-inducible protein-10, monocyte chemoattractant peptides (MCP)-122-24) and macrophage inflammatory protein-2 appear in the kidney within 2-5 days of ischemia-reperfusion injury (IRI). This late appearance suggests that macrophages may participate in the repair process after IRI. However, the recent findings by Day et al. that macrophages play a critical role in mediating the full extent of IRI are significant for the role of macrophages in IRI25). In addition, Jo et al. reported that macrophages contribute to the initiation of ischemic ARF in rats26). They suggested that macrophages were an important mediator in the initiation and extension of ischemia/reperfusion injury, and those strategies that limit the initial macrophage infiltration or activation can be useful for treating acute renal failure26).
The present study found that there was an increased number of CD 11b-positive cells from 2 h after IRI, and that macrophage infiltration peaked at 16-24 h. This finding is consistent with the recent report that ED-1 cell infiltration began to increase by 4 h after IRI, and it had markedly increased by 24 h. Our present study found that renal function was impaired by 16 h after IRI, and this impairment was maximal at 24 h. Despite the rapid and significant recruitment of macrophages from 2-8 h after IRI in our study, this might not be sufficient to induce the deterioration of renal function during that time.
The term acute tubular necrosis (ATN) is a misnomer because frank tubule cell necrosis is rarely encountered in human ARF. Necrosis is inconspicuous and it is restricted to the highly susceptible outer medullary regions6). Even under normal conditions, the outer medullary region exists on a hypoxic precipice as a result of low blood flow and the countercurrent exchange of oxygen, although paradoxically, this outer medullary region houses nephron segments with very high-energy requirements (e.g., the S3 segment of the proximal tubule and medullary thick ascending limb of Henle's loop)6). As such, the current study investigated the location of macrophage infiltration after ischemic insult. As might be expected, the infiltrating macrophages were mainly found in the outer medullary region.
The extension phase of ischemic ARF involves continued reduction in renal perfusion, the ongoing hypoxia and the inflammatory processes that occur during reperfusion, and this all contributes to continued tubular cell injury. Endothelial-leukocyte interactions mediated through complementary adhesion molecules on the endothelial cells and leukocytes play a key role in the local accumulation of leukocytes, such as macrophages, during the extension phase of ischemic ARF. Ischemic ARF leads to an increased endothelial expression of a variety of adhesion molecules, including ICAM-1, P-selectin and E-selectin, which all promote endothelial-leukocyte interaction6). A great deal of attention has been directed towards the peritubular capillary, and especially those peritubular capillaries in the outer medullary region6). The interaction between adhesion molecules on the inflamed endothelium and inflammatory cells such as macrophages results in the striking vascular congestion and hypoperfusion of the outer medulla, and this persists even though the cortical blood flow improves during reperfusion after an ischemic insult2, 3). Infiltrating macrophages can also induce direct cytotoxic effects on the renal epithelial cells25). According to our findings, we can suggest that macrophage infiltration plays a pathogenic role in ischemic ARF, and especially in the extension phase of ARF.
The present study found that clodronate administration prevented the ischemia-induced increase of the serum creatinine levels, and this indicated that these effects were specifically the result of macrophage depletion. Other investigators have shown that in an experimental model of uveitis, liposomal dichloromethylene-diphosphonate administration depleted the macrophage component and it prevented leukocyte-endothelial interactions, thus providing evidence for the central role of macrophages in this form of inflammation27). In arterial injury, macrophage depletion with using clodronate reduced the inflammation that was involved in neointimal hyperplasia28). Those studies provide further strong evidence for the role of macrophages in inflammation and renal ischemic ARF. In addition, two recent studies indicated that macrophage depletion could protect against ischemic injury25, 26).
In summary, the present study of ischemic ARF found that there was a significant infiltration of macrophages from 2 h after ischemic ARF, that this infiltration peaked at 16-24 h, and that the macrophages were mostly in the outer medullary region. Our study found that macrophage depletion using clodronate was protective against ischemic ARF.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





