Nonspecific Interstitial Pneumonia/Fibrosis: Clinical Manifestations, Histologic and Radiologic Features
Article information
Abstract
Objectives
Customarily used classification of IPF did not satisfy a sizable group of patients with IPF that in the past had been lumped with UIP and now currently has begun to be recognized as nonspecific interstitial pneumonia/fibrosis (NIP). There are few reports about the clinical features of NIP.
Methods
The pathologic slides of 66 patients having open lung biopsy (OLB) for the differential diagnosis of interstitial lung diseases (ILD) from 1984 to 1995 were reviewed. Seven cases were confirmed as NIP. The clinical record, pulmonay function tests (PFT), chest PA and HRCT were reviewed and analysed retrospectively.
Results
Six patients with NIP were female. The median age was 56 years. Mean duration of symptoms was 4 months. Five patients had systemic flu-like symptoms. the most common respiratory symptom was gradual dyspnea Two patients revealed a mild degree of anemia Four cases had leukocytois of more than 10,000/mm3. ESR and CRP O.K. elevated in all measured cases. Anti-nuclear antibody (ANA) was positive in three of six patients and ds-DNA antibody was positive in one of six patients Restrictive pattern of PFT was predominant. Diffusion capacity of carbonmonoxide (DLCO) decreased markedly. In bronchoalveolar lavage (BAL), total cell counts elevated about three times of normal value. On differntial counts of BAL cells, lymphocytes, neutrophils and eosinophils were higher than those of normal controls. The prominent finding of chest radiology was bilaterally patchy opacifications in parenchyme of lower lung zones. On HRCT, bilaterally patchy areas of ground-glass attenuation and/or areas of alveolar consolidation were commonly shown. The number of pathologic type was one case of group I, four cases of group II and two cases of group III. The average period from diagnosis to the last follow-up was 24.8 months. Five patients were clinically recovered to the previously well-being state.
Conclusion
Patients with NIP had different clinical features from UIP, AIP and DIP. They also had characteristic findings of radiology and their prognosis seems to be better than UIP.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a group of diseases that is characterized by a progressive fibrosing inflammation of pulmonary interstitium and air spaces. Hamman and Rich reported the clinical and pathologic findings of four cases, who expired due to a fulminant pulmonary fibrosis with rapidly progessing course in 19351). Thereafter, many cases of diffuse interstitial pneumonias with chronic courses have been reported. Although the terminology has been used inconsistently, IPF represents the interstitial penumonias without any evidence of underlying causes or other disease.
In 1967, Liebow and carrington classified interstitial pneumonias into five subcategories by pathologic findings, usual interstitial pneumonia (UIP), desquamative interstitial pneumonia (DIP), bronchiolits with interstitial pneumonia (BIP), lymphocytic interstitial pneumonia (LIP) and giant cell interstitial pneumonia (GIP)2,3). In 1986, Katzenstein4) identified acute interstitial pneumonia (AIP) as a new subcategory of fibroblasts is a main component in AIP, whereas the dense deposition of collagens is prominent in UPI. Katzenstein also indentified a sizable group of patients who cannot be subcategorized pathologically into one of above three entities of IPF and termed nonspecific interstitial pneumonia/fibrosis (NIP) in 19945).
NIP is distinguished from UIP by a temporal uniformity of interstitial inflammations and/or fibrosis. The clinical feature to differentiate NIP from UIP is that the former seems to have a better prognosis than the latter. NIP is also distinguished from DIP or AIP by a more prominent interstitial inflammation or fibrosis. Since Katzenstein’s report5), NIP has been accepted and used as an unique entity of IPF. However, there have been few reports about the clinical features of NIP till now.
We firstly reported clinical features of three cases at the 77th Conference on Tuberculosis and Respiratory Diseases of Korea in 19936). Because of the shortage of clincal data so far, we summarized the clinical features, radiologic and pathologic findings of seven patients with NIP.
MATERIALS AND METHODS
1. Study Population
Study population was composed of 66 patients having open lung biopsy (OLB) for the differential diagnosis of interstitial lung diseases (ILD) from 1984) to 1995. After clinical evaluations including pulmonay function tests (PET) and HRCT were taken, OLB was performed at the determined sites in consideration of chest PA and/or HRCT findings. For pathologic proof, all pathologic specimens were reviewed by two pathologists in our hospital. The cases of interstitial pneumonia were consulted outside, mainly with Professor Kitaichi in Kyoto University, when they were not compatible with UIP, DIP or AIP. Six cases of NIP were pathologically confirmed by Professor Kitaichi and one cases of NIP by Dr. Katzenstein in the State University of New York, New York.
NIP is characterized by a variable degree of intersitial inflammation with or without fibrosis. However, it appeared temporally uniform within each case and areas of honeycombing were not found. Histologic features for a specific category of interstitial pneumonia were not present. These cases were divided into three groups according to Katzenstein’s criteria5). Group I was characterized by a cellular interstitial pneumonia with littly fibrosis. The interstitial infiltrate was minimal to mild and consisted of small lymphocytes (Fig. 1). There were patch involvement and accentuation around bronchioles. Group II was characterized by a cellular interstitial pneumonia with significant fibrosis (Fig. 2). The alveolar septa were widened by cellular infiltrations, which consisted of lymphocytes, plasma cells and a few eosinophils. An area of germinal center formation was noted. There was a marked hyperplasia of alveolar lining cells. The fibroblasts were composed of mature collagen and proliferating fibroblasts. Large numbers of intraalveolar macrophages and fibroblast foci were found. Group III was characterized by architectural derangement due to dense interstitial collagenous fibrosis and less degree of inflammation (Fig. 3). This process was diffuse in one case and patchy in the other case. The alveolar septa were markedly thickened, resulting in narrowing of air spaces. The alveolar lining cells were hyperplastic and alveolar spaces were filled with alveolar macrophages. Areas of patchy interstitial infiltrate were noted, but their degree was mininal compared to that of group II.
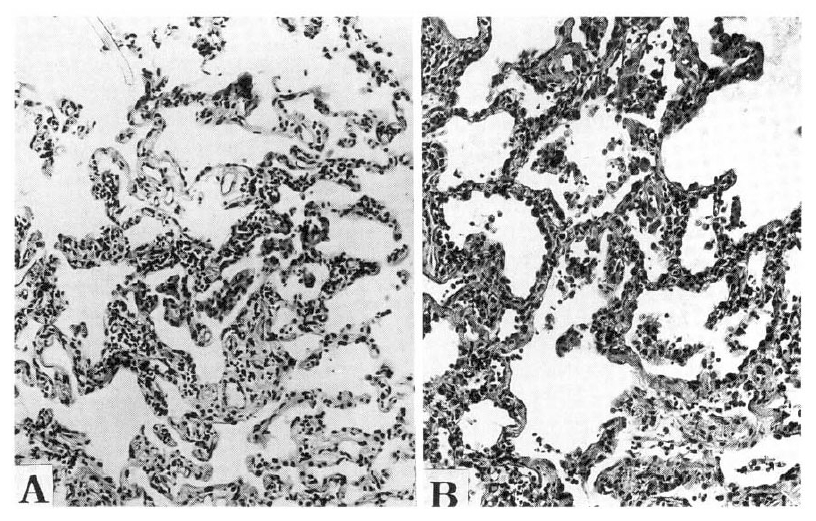
Light microscopic findings of Group I. Group I was characterized by a cellular interstitial pneumonia with little fibrosis. The interstitial infiltrate was minimal to mild and consisted of small lymphocytes. Involvement was patchy and was accentuated around bronchioles (Hematoxyline eosin stain, A figure: 40X, B figure; 100X)
Light microscopic findings of Group II. Group II was characterized by a cellular interstitial pneumonia with significant fibrosis. The alveolar septum was widened by cellular infiltrations, which consisted of lymphocytes, plasma cells snd a few eosinophils. An area of germinal center formation was noted. There was a marked hyperplasia of alveolar lining cells. The fibroblasts were composed of mature collagen and proliferating fibroblsts. Large numbers of intraalveolar macrophages and fibroblast foci were found (Hematoxyline eosin stain, A figure; 40X, B figure; 100X).

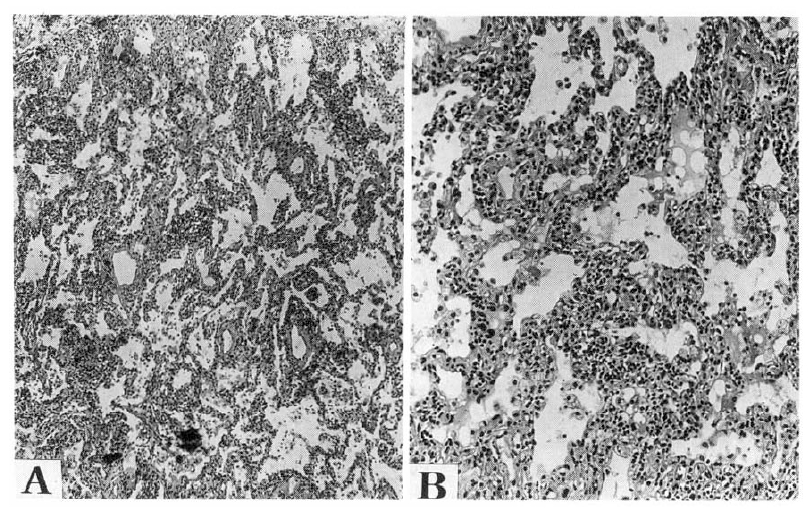
Light microscopic findings of Group III. Group III was characterized by architectural derangement due to dense interstitial collagenous fibrosis and lesser degree of inflammation. These processes were diffuse or patchy. The alveolar septum was markedly thickened, resulting in narrowing of air spaces. The aveolar lining cells were hyperplastic and alveolar spaces were filled with alveolar macrophages (hematoxyline eosin stain, A figure; 40X, B figure; 100X).
2. Clinical Features and Manifestation
Chart of seven patients were reviewed. A standardized history taking and physical examinations were performed by the charged physicians. The standardized history included duration of respiratory symtoms, past medical history, family history, smoking history, medication history and occupational history with exposures to organic or inorganic dusts or toxic fumes. Clinical histories were reviewed to identify any possible underlying diseases or other potential causes of pulmonary abnormalities. Respiratory questionnaire included dyspnea, cough, sputum, chest tightness and chest pain Rales, rhonchi and wheezing were also identified as respiratory signs.
3. Routine and Special Blood Tests
Routine blood tests included CBC, liver function tests and kidney function tests. Serologic and immunologic tests included ESR, C-reactive proten (CRP), anti-nuclear antibody (ANA), rheumtoid factor (RF), double stranded-DNA (ds-DNA) antibody, LE cell test, immunoglobulin (Ig) G, A, M and complements (C3, C4).
4. Measurement of Pulmonary Function Tests
Prior to OLB, forced vital capacity (FVC), forced expiratory volume at one second (FEVI), total lung capacity (TLC) and DLCO were measured with pulmonary function analyzer ERS-1500 (Fukuda-Sankyo, Tokyo). TLC was measured with a heliumdilution method. All data were expressed as percentages of predicted values. Diffusion capacity for carbon monoxide (DLCO) was measured by the single-breath method and expressed as a percentage of the predicted values.
5. Procedures of Bronchoalveolar Lavages
Six Patients with NIP underwent bronchoalveolar lavage (BAL). One patient was excluded due to severe dyspnea. BAL was done as usual. The sites of BAL were decided in consideration of HRCT findings. Four times’ instillation of 50 ml sterile normal saline and gentle suction with negative pressure below 50 mmHg was done. Total cell count were calculated with a hemocytometer (American opics, USA). Cell pellets were sparated by centirifugation at 500 g for 5 min. Differential count of 500 cells was done on the slides prepared by cytocentrifuge and Diff-Quik stain.
6. Clinical Course and Follow-up
PET and HRCT performed again at the 2nd week after OLB and then steroid a were begun to be administered if the patients did not need urgently tereatment. Prednisolone was given orally in a single dose with 1 mg/kg of body weight for 2 weeks to 4 weeks and then was tapered 10 mg every one or two weeks, if patients showed clinical response of improvement. At 4th week, 3rd, 6th, and 12th month after treatment with steroids, follow-up measurements of PET and HRCT were performed.
The clinical response was analysed by a subjective improvement of dyspnea and by the objective evaluation of PET and HRCT. The physiologic improvement was defined when the increment of FVC was more than 10%, or that of DLCO or TLC was greater than 20% compared to those of initial values. The stationary state was defined when the changes of FVC was less than 10%, or that DLCO or TLC was less than 20%.
7. Statistics
SPSS/PC program (Chicago, Illinois, USA) was used. The differences between groups BAL cell analysis were compared with the nonparametric Mann-Whitney U test. The difference was considered significant when p value was less than 0.05. All results were expressed as mean ±SE.
RESULTS
1. History, Symptoms and Signs of the Study population
Among the sixty-six patients having OLB for the differential diagnosis of interstitial lung disease (ILD) from 1984 to 1994, thirty-five patients were proved as IPF and 10 patients as collagen vascular disease-associated pulmonary fibrosis (CVD-PF). Among IPF, seven patients were diegnosed as NIP. The first case was diagnosed in June, 1992 and the lat one in May, 1994.
Female was the predominant gender of the patients with NIP: six patients were female and one patient was male. The mean age of the patients was 56 years and ranged from 43 to 69 years. One male patient was a current smoker, and the others were never-smokers. As for the undrlying and associated medical conditions, one patient had diabetes mellitus for about six years, an other one had mild bronchial asthma for three years and another patient had hypertension for three years.
As an initial symptom, five patients had systemic flu-like discomfort such as fever, myalgia and fatigue for a few days. Two patients had elevated body temperature (37.5°C and 37.6°C). Mean duration of symptoms was 4 months before diagnosis. They were followed up for more than 12 months, except one patient who was followed up for 1 month after discharge. However, she has been contacted by telephone till now at 16 months after discharge (Table 1).
Demographic Features of Patients with Non-Sepcific Interstitial Pnemonitis
The most common respiratory symptom was dyspnea, which all of the seven patients complained of The median grade of dyspnea was about 10 according to CRP classifications7). Four patients complained of cough and chest tightness, three patients complained of sputum, not purulent but mucoid, and two patients had chest pain. On physical examinations, rale was audible in all patients. One patient had wheeze on expiation (Table 2).

Respiratory Symptoms and Signs of Patients with NIP
2. Routine Blood Examinations and Serologic Data
On complete blood counts, two patients had a mild degree of anemia: 10.2% and 10.5% Hb. Peripheral blood morphology, RBC index, the level of serum iron. TIBC and ferritin indicated that their anemias were due to a complication of chronic diseases. In four cases, WBC elevated more than 10,000/mm3 (22,500/mm3, 19,400/mm3, 12,300/mm3). Differeential counts were within normal limit in all patients, except one patient who had eosinophilia with 9%. Platelet count was normal in all patients. One patient had and elevation of SGT (45.61 IU/ml), SGPT (36.81 IU/ml) and alkaline phosphatase (122.61 IU/ml). Kidney function tests were normal in all patient (Table 3).

Profiles of Prlmonary Function Tests, in Patients with NIP
ESR was elevated significantly in all patients with a mean of 41±4 mm/hr rage from 24 mm to 57 mm/hr. All of four measured cases had more than one positive value of one of six patients. LE cell was negative in all cases (Table 3). One patient had a positive ds-DNA antibody, but she did not have the other manifestations of SLE. The levels of IgG (1883±193 mg/dl), IgA (321±93 mg/dl), IgM (339±56 mg/dl), C3 (116.3±35 mg/dl) and C4 (31.2±18.0mg/dl) were within normal ranges in the three measured patients.
3. Pulmonary Function Tests
FVC decreased in six patients. Three patients showed a severe degree of restriction less than 50% of predicted value, two showed a moderated degree of restriction betwee 51% and 65% of predicted value and one showed a mild degree of restriction between 6% and 75% of predicted value. FEV1/FVC was normal in all cases. TLC ws measured in 6 cases. One male patient had normal ranges. of TLC. In five cases, TLC was decreased from mild to severe degree. DLCO decreased markedly in two of three measured patients, and the other patients could not perform PET adequately because of difficulty in breath holding (Table 4).

Inflamatory Cell Profiles of Bronchoalveolar Lavage fluid in Patients with NIP
4. Analysis of Broncholveolar Lavage Cells
BAL was performed in 6 cases. Profiles of BAL cells were compared with those of normal controls which were published previously8). The mean recovery rate was a mean of 43.6±4.6%, which was lower than that of the normal controls (p<0.05).
Total cell count was a mean number of 57.5±17.53×106. which was about three times of normal value (p<0.01). On differential counts, the percentage of lymphocytes were 36.5±9.10% and eosinophils 4.8±2.59%. In the patients with NIP, there was a marked increase in the percentages of lymphocytes (p<0.01), neutrophils (p<0.01) and eosinophils (p<0.05) when compared to those of normal controls (Table 5).

Numbers of Abnormal Histologic Findings in Patients with NIP
5. Chest X-ray and HRCT Findings
A prominent finding of chest radiology was a patchy opacification of parenchyme in six patients. It bilaterally involved the middle (n=6) and lower (n=6) lung zones more than the upper lung zone (n=2) (Fig. 4). In one patient, the parenchymal abnormality was combined with opacifications and reticular densites. In the remaining one patient, no abnormalities were noted on chest radiograhp, but HRCT, obtained two days later, depicted pathy areas of ground-glass attenuation.
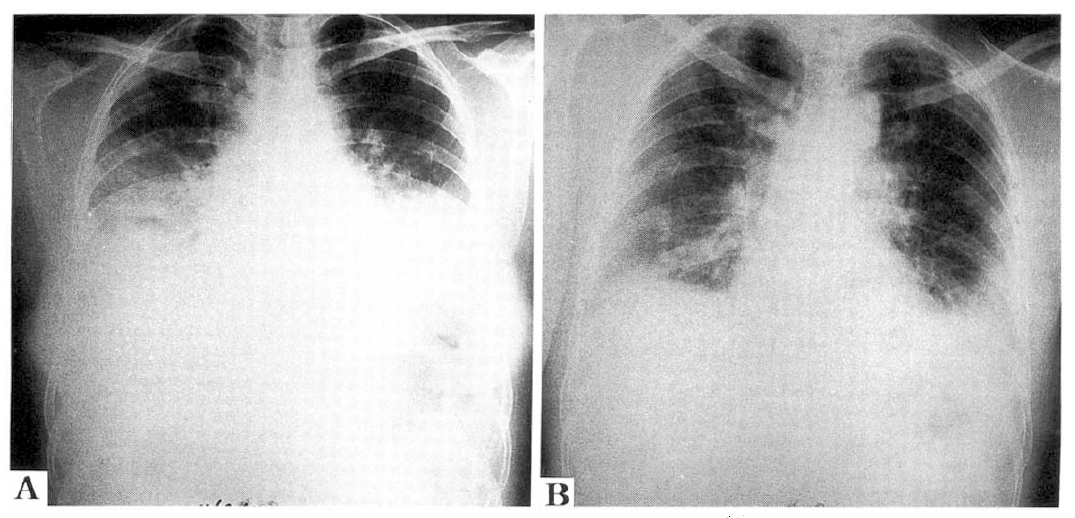
Chest PA findings of NIP. Chest PA showed a bilaterally patchy opacfication of parenchyme in the middle and lower lung zones. The parenchymal abnormality was combined with opacifications and reticular densities (left figure). Follow-up chest PA showed much improvement of patchy opacification. However, patchy areas of ground-glass attenuation were still present on both lower lung zones (right figure).
On initial HRCT scan, the most common findings were bilaterally patchy areas of ground-glass attenuation alone and/or areas of alveolar consolidation in five patients, without central or subpleural predominancy (Fig. 5). Irregular lines were present for two patients (29%) without honey-hcombing or traction bronchioloectasia on initial or follow-up HRCT scans. Enlarged lymph node was observed in two patients. Pleural effusin was not present in any patient. Follow-up CT scan revealed that the initial abnormalities had resolved completely in three patients within 13 months. In the other three patients, the overall extent of parenchymal abnormalities had improved.
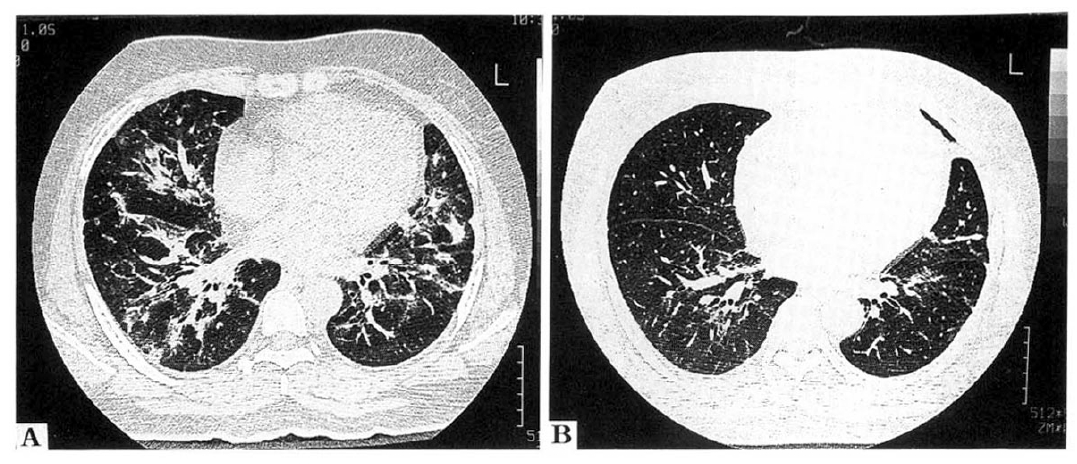
HRCT findings of NIP. An initial HRCT scan showed bilaterally patchy areas of ground-glass attenation alone and/or areas of alveolar consolidation with central or subpleural predominancy. Irregular lines were present without honeycombing or traction bronchioloectasia (left figure). Follow-up CT scan revealed that the initial abnormalities had resolved completely 13 months after treatment (right figure).
6. Pathologic Findings
Our cases were divided into three groups a ccording to Katzenstein’s criteria5). Salient pathologic findings were summarized in Table 6. Only one case blonged to Group I, four cases were catergorized as Group II and two cases were in Group III.
Clinical Courses and Responsiveness to Steroid Treatment
7. Clinical courses and Follow-up Evaluations
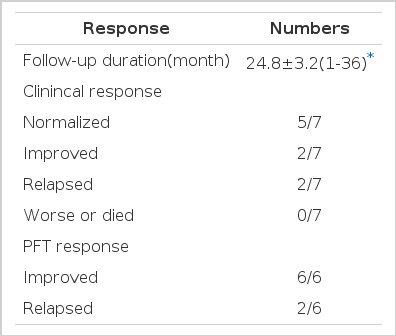
Corticosteroid was trid on 6 patients. Because one female patient showed subjective improvement during two weeks after OLB, she did not receive steroids and was followed up till now without symptomatic aggravation. The average period from diagnosis to the last follow-up was 24.8 months. Six patients were regularly followed up form 12 months to 36 months. One patient was not followed up at our hospital after first month of treatment with steroids. By correspondence, dyspnea was revealed to the be makedly improved, but she was excluded from the data analysis.
All patients felt improvement dyspnea within 2 weeks to 8 weeks spontaneously or by trial with steroids. Five patients completely recovered to the previously well-being state before the illness within one to five months later. One female patient felt dyspnea to be gradually improved during 9 months’ trial with steroids, but not to disappear completely. The recovery of subjective symptoms took a mean duration of 4.2 months (Fig. 6). All of he six patients showed improvement of PET during follow-up. FVC and TLC were recovered to normal values spontaneously in one patient and by treatment with steroids in five patients (Fig. 7). The recovery of FVC took a mean duration of 6.3 months. However, DLCO has remained lower than the predicted value during follow-up. In two cases dyspnea had relapsed 4 months and 6 months after the discontinuation of steroids. Three patients remained asymtomatic and had normal FVC and TLC at 4th, 12th and 15th months after the discontinuation of steoids. One case, who needed corticosteroid, still remained with sub-normal lung functions at 30th month of follow-up period. She had a remarkable foci of BOOP on the pathologic findings and was classified as Group 3.
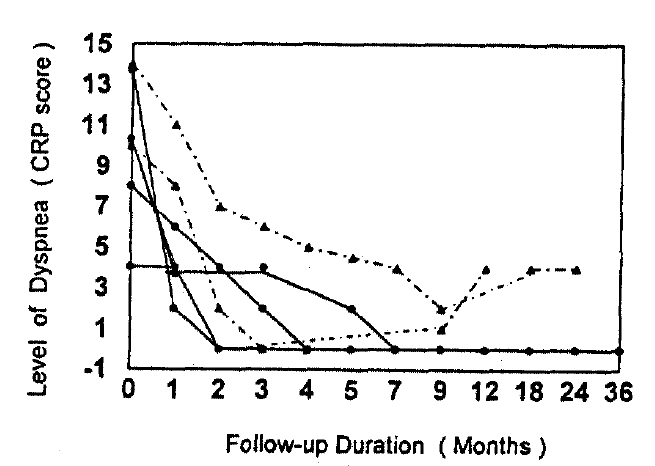
Changes of dyspnea score in 6 patients with NIP. The solid line represented the patients responding to steroid treatment and without recurrence of dyspnea during follow-up. The dotted line represented the patients with recurrence of dyspnea after withdrawal of steroids.

Changes of FVC in 6 patients with NIP. The solid line represented the patients responding to treatment and without aggravation of FVC during follow-up. The dotted line represented the patients with aggravation of FVC after withdrawal of steroids
DISCUSSION
Since Liebow’ classification2) for IPF, UIP is known to be the most common from of IPF. It composes the bulk of cases classified under the clinical category of IPF. It is a chronically progressive course with a mortality of more than 60% within 5 years1,6,12).
DIP, as a less common form than UIP, showed very good prognosis with 5 years’ mortality of 25%9).
However, there remains a sizable group of interstital pneumonias termed NIP wiich cannot be subcategorized into the above well-defined categories. Yousem et al.12) and Tazelaar et al.13) described a chronic interstitial pneumonia distinct from UIP that they termed cellular interstitial pneumonia. This lesion likely corresponds to cases of NIP in Katzenstenin’s and our study.
NIP, reported by Katzenstein5), is distinguished from UIP by temporal uniformity of interstitial changes and patchy infiltrations of intraalveolar macrophages. These pathologic differences are also prominent in radiologic findings characterized by bilaterally pathy areas of infiltrations14).
The frequency of NIP in our patients was 7 patients (20%) of the total 35 patients with IPF diagnosed on biopsy from 1984 to 1995 in our hospital. However, the time in which our patients were diagnosed NIP is interesting because they showed a limited period from June 1992 to July 1995. We explain the reason why there was no case of NIP before 1992. It is possible that new viral or environmental agents played or triggered a role in the development of NIP. We did not find any abnormal history of mediction, occupation and family.
The etiology of NIP is usually unknown as is that of IPF. Kazenstein5) considered a number of possible etiologic factors for NIP as follows: underlying connective tissue disease, exposure to pet parrot, canary, wood stove, grain dust, coal and ash. The occupations of patients were very variied, including farmer, paper factory worker, worker for a veterinarian and garment worker. In Katzenstein’s study, 10 patients (16%) had an associated connective tissue disease. In our patients, one female patient showed positive ds-DNA antibody of slightly above upper normal limit, but she was not compatible with the criteria of SLE. The other patients did not have any of the above possible, underlying diseases.
According to the Kyoto University’s data155, they had 4 cases (5%) of NIP report of the 88 patients with idiopathic interstitial pneumonia. The ratio of NIP to UIP was 4 to 73 cases. The incidence of NIP seems to be relatively higher in Korea than Japan. Katzenstein5) reported that female is slightly higher than male (38.26). In our patients, female gender was very perdominant (F:M ratio of 6:1). The average age was 46 (range from 9 to 78) and patients younger than 20 years of age were 5 in that report. The average age of our cases was slightly older than that of Katzenstein’s study. This may be due to lack of open lung biosy in patients in our study.
All of our patient had the subacute and chronic pesentation of symptoms less than one year’s duration as in almost all cases in Katzenstein’s report. All our patients had systemic flu-like symptoms at the onset of NIP, but two patients had only elevated body temperature. The incidence of fever is comparative to that of Katzenstein’s study (14 of 64 patients). The most common presenting symptom in our study was a gradually increasing dyspnea, which are comparative to the incidences study.
On laboratory, our patients showed the elevated level of WBC counts and ESR and positive reaction of CRP in 5 patients. These are thought to be due to nonspecific inflammations and not due to infections, Although the number was limited, the levels of IgG, IgA, IgM, C3 and C4 in all measured patients were within normal ranges. This may suggest that immune complex does not present remarkable laboratory abnormalities.
Our patients revealed a similar pattern of PFT as observed in other patients with interstitial pneumonias, such as a dominant restrictive pattern and minimal obstructive pattern16,17), Residual volume and functional respiratory capacity were increased in one male patient and this phenomenon was considered to be due to smoking. DLCO remained lower than the predicted value during follow-up even after lung volumes returned to normal vlues. This suggfests that subclinical inflammation in alvoli is persistent even in asymptomatic patients after treatment with steroids.
In IPF, it was known that the total number of inflammatory cells in BAL fluid increases in proportion to the activities of disease and steroid responsiveness was good if lymphocytes were predominant18,19). In our series, the total cell count was markedly elevated. On differential count, lymphocyte and neutrophil were the predominant inflammatory cells. These can indicate a good prognosis of NIP.
Our patients presented bilateral patchy parenchmal infilrates, frequetly on the middle and lower lung field on the chest radiographs which was already noticed in Katzenstein’s report. On HRCT scan, bilateral patchy areas of ground-glass attenuation alone or areas of alveolar consolidation were the most common findings which are distinguished from UIP having irregular linear areas of attenuation leading to the reticular pattern and honeycombing predominantly involving the subpleural regions and lower zones20) and DIP having the distribution of abnormalities in subpleural lower lung zones21).
The majority of patients with UIP were treated with steroids, cyclophosphamide or azathioprine22,23) but the response was not good. Our patients showed good response not only on the clinical course but also on PET. With the mean duration of 24 months of follow-up, improvement of dyspnea was noted in all six measured patients wth NIP and five patients were completely recovered to the previously wellbeing states before the illness. Response of PET to the therapy revealed improvements in all six measured patients. Thus, the prognosis seems to be better than UIP. This finding was also noticed in Katzensteins’ report5). Only 5% of patients died of progressive lung disease, whereas 83% patients either recovered or were alive with the disease. Importantly, complete recovery was possible even in the case with the more severe fibrosis of Group III.
Although the number of our study patients with NIP was limied, we conclude that NIP presents a distinct entity of IPF with different clinical features, rediology and pathology from other forms of IPF.