Polymorphism of Glucokinase Gene in Non-Insulin Dependent Diabetes Mellitus
Article information
Abstract
Several lines of evidence suggest a strong genetic component to NIDDM.
To clarify the role of glucokinase gene in the development of NIDDM, restriction fragment length polymorphism (RFLP) of glucokinase gene and 3′ microsatellite polymorphism analyses by polymerase chain reaction-single strand conformational polymorphism (PCR-SSCP) were performed in NIDDM and control subjects.
Compared to NIDDM with 1.3 kb allele/Pvu I digestion of glucokinase, 10% of NIDDM did not demonstrate 1.3 kb allele and these patients were charcterized by increased insulin secretion. In 3′ microsatellite polymorphism analysis, autoradiography of PCR products revealed three different alleles, including Z, Z+2 and Z+4. Z was the most common allele in both NIDDM and nondiabetic controls. There was no significant allele associated with NIDDM. Frequency of the homozygote Z/Z genotype was significantly lower in NIDDM subjects (16.7%) compared to normal control (46.7%) (p<0.05).
There was no difference in clinical findings according to 3′ microsatellite genotypes in NIDDM. These data suggest that there does not appear to be a significant glucokinase allele associated with NIDDM but Z/Z genotype may play a suppressive role in the pathogenesis of a certain type of NIDDM in Korea. Further studies may be required to identify the molecular basis of this association.
INTRODUCTION
Despite a decade of investigation, the pathogenic mechanism of non-insulin dependent diabetes mellitus (NIDDM) remains one of the unsolved mysteries. Under normal circumstances, glucose homeostasis is a balance between the glucose production by the liver and glucose clearance into the peripheral tissues, primarily muscle. The beta cell is a regulator of this system; insulin release is constantly adjusted so that glucose production and clearance change in ways that maintain normoglycemia. In NIDDM, all three components of the system are defective: the clearance of glucose from the circulation falls, insulin release after meal is slowed and the glucose production rises1).
Several lines of evidence suggest a strong genetic component to NIDDM. Marked differences in prevalence have been noted among various racial groups. Also, studies of identical twins with NIDDM have clearly shown that the disease is genetic, although the pattern of inheritance has not been defined as yet2).
The search for diabetic genes in NIDDM has intensified as of the late 1990’s. Attention focused first on finding mutations in persons with rare syndromes of diabetes, in whom the mode of inheritance suggested single gene defects. Mutant insulins have been found in families with extreme hyperinsulinemia or hyperproinsulinemia. More than 40 different mutations in the insulin receptor have been identified in patients with insulin resistance syndromes. Most recently, mutations in the key regulatory enzyme of the beta cell, the ‘glucose sensor’ glucokinase, have been discovered in families with maturity onset diabetes of the young (MODY), a form of the disease distinct from typical NIDDM in that it has an onset early in life and is inherited in an autosomal dominant manner3). Attention has now shifted to finding mutations in the common form of NIDDM. Candidated genes are chosen on the basis of abnormalities of metabokism that are known to occur in this disorder.
Glucokinase is the key enzyme of the regulation of glucose homeostais. Glucokinase gene is expressed exclusively in liver and pancreatic islet beta cells. In hepatocyte, this enzyme plays a role in facilitating the uptake and metabolism of glucose and in the pancreatic beta cell, in glucose sensing and in initiating glucose-induced insulin secretion4). Thus, defects in the glucokinase gene may contribute to the development of NIDDM.
Recently, the glucokinase gene structure has been characterized and was found to contain two different transcription control regions. One region regulates transcription of the gene in the liver, whereas the other region, which lies at least 20 kilobases further upstream, controls transcription in the pancreatic beta cell. The finding of two different transcription control regions in a single glucokinase gene provides a genetic basis for the tissue specific differential regulation of glucokinase. And the possibility of a feedback loop involving hepatic and beta cell glucokinase has been proposed. Hepatic glucokinase, which is stimulated by an increase in the plasma insulin concentration, affects the rate of glucose usage by the liver, thereby helping to lower the plasma glucose concentration. On the other hand, increased plasma glucose would induce expression of glucokinase in the beta cell, this causing increased beta cell glycolysis and greater insulin secretion4).
To clarify the role of glucokinase gene in the development of NIDDM, restriction fragment length polymorphism (RFLP) of glucokinase gene and 3′ microsatellite polymorphism analyses were performed.
MATERIALS AND METHODS
1. General Procedure
Peripheral blood leukocytes were obtained from 30 diabetic patients according to the criteria of National Diabetes Data Group and 30 healthy controls. In patients with NIDDM, serum insulin, c-peptide and 24 hour urine c-peptide were measured by radioimmunoassay method.
Basic molecular laboratory procedures were performed according to standard protocols of this laboratory.
2. Southern Blot Analysis of Human Genomic DNA
Glucokinase gene RFLP analysis was performed as in previously reported methods5). In brief, genomic DNA were extracted from blood leukocytes by phenol-chloroform methods and were digested by Pvu II restriction enzyme. The DNA fragments were separated according to the size in an agarose gel electrophoresis and transfered to nitrocellulose filter by the method of Southern transfer. The amplified and purified rat glucokinase whole cDNA (pGK,Z9: kindly donated by Dr. Magnuson MA, Vanderbilt University, Nashville Tennessee) was radiolabelled for use as a DNA probe. After hybridization of the nitrocelluose filter with radiolabelled DNA probe, exposure to X-ray film was performed to obtain autoradiogram.
3. Microsatellite Polymorphism (Fig. 1)

PCR method for analysis of the polymorphic (CA)n repeat region.
Following experiment was done to identify the association between NIDDM and microsatellite polymorphism of glucokinase gene. PCR-SSCP (polymerase chain reaction-single strand conformational polymorphism) assay was done for 8 kb downstream microsatellite region.
A set of oligonucleotide primers flanking the microsatellite region was used for PCR amplification. Sense primer was 5′-end labeled for 45 min at 37°C in a 25- μl reaction containing 25 pmol primer. T4 polynucleotide kinase. Each PCR assay contained 250 ng intact genomic DNA, 10 pmol antisense primer, 9 pmol unlabeled and 1 pmol 32P end-labeled sense primer, 200 μM each dNTP, 1.5 mM MgCl2, and 1 unit Taq polymerase in 50 μl reaction volume with the buffer supplied in the Gene AMP kit (Perkin-Elmer-Cetus, Norwalk, CT). Sample was processed through initial denaturation for 3 min at 95°C, 30 cycles of amplification, which consisted of 50 sec at 95°C (denaturation) and 50 sec at 66°C (annealing-extension) and final elongation at 72°C for 5 min in Perkin-Elmer-Cetus thermal cycler. After being denatured at 75°C for 2 min in formamide stop solution (50% formamide and 10 mM EDTA), aliquots of amplified samples were run in alternate lanes on a standard DNA sequencing gel containing 6% acrylamide and 7 M urea in 27 mM Tris-borate, 0.6 mM EDTA buffer, for 2.5 h at 70 w. Gels were then fixed, dried and processed for autoradiography.
4. Statistical Analysis
Differences between groups in quantitative variables were evaluated by unpaired t test. Association between allelic and genotypic frequencies in diabetic and nondiabetic subjects were assessed with χ2 test of independence.
RESULTS
1. Glucokinase Gene RFLP
In NIDDM patients, negative 1.3 kb allele was more common than in normal subjects (Table 1). Clinical manifestations according to the RFLP of glucokinase gene, age at onset, fasting blood sugar, fasting serum insulin and c-peptide level were not different between the 1.3 kb allele negative and positive group. However 24 hr urine c-peptide excretion rate was significantly more elevated in the 1.3 kb negative group than in the 1.3 kb positive group. Also, insulin and c-peptide response to oral glucose was more increased in the 1.3 kb negative group than in the 1.3 kb positive group (Table 2).

Frequency of 1.3 kb Allele of Glucokinase Gene

Clinical Manifestations According to Glucokinase Gene RFLP in NIDDM
These result may suggest the following hypothesis. The NIDDM patient of negative 1.3 kb allele of glucokinase gene may have the defect of hepatic glucokinase gene.
2. Microsatellite Polymorphism
Allelic and genotypic frequencies of the dinucleotide repeats at the glucokinase locus are shown in table 3 and table 4. Autoradiography of PCR products of normal control revealed three different alleles including Z (73.3%), Z+2 (16.7%) and Z+4 (10.0%)(Fig. 2). In NIDDM, frequencies of microsatellite repeat alleles were as follows; Z, 58.3%, Z+2, 25.0%, Z+4, 16.7%. Z was the most common allele in both NIDDM and nondiabetic controls. There was no significant allele associated with NIDDM.

Allelic Frequencies of Microsatellite Polymorphic Marker for Glucokinase Gene

Genotypic Frequencies of Microsatellite Polymorphic Marker for Glucokinase Gene

Alleles PCR amplified from 3′ microsatellite region in NIDDM.
Genotype of microsatellite polymorphic markers was observed, including Z/Z, Z/Z+2, Z+4 (Table 4). In normal control, Z/Z (46.7%) was the most common genotype. Compared to normal control, the frequency of the Z/Z genotype (16.7%) was significantly lower in NIDDM subjects (P<0.05). No differences were noted between the frequency of the other genotypes in the control group and NIDDM (Z/Z+2, 33.3% vs 50.0%, Z/Z+4, 20.0%, vs 33.3%).
There was no difference in clinical findings according to 3′ microsatellite genotypes in NIDDM (Table 5).
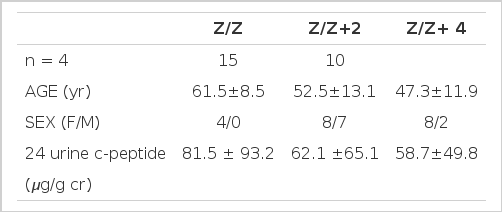
Clinical Findings of NIDDM According to Genotype
DISCUSSION
NIDDM is one of the most common metabolic diseases that affects 5% of the world population, yet its causes have remained largely unknown. The contribution of heredity to the development of NIDDM has been recognized for many years6,7). A number of different genetic mechanisms that contribute to the susceptibility of NIDDM have been investigated in some racial groups using RFLPs, including insulin8,9), insulin receptor genes10), mitochondria11,12), glucose transporter isoforms13,14), and various apolipoprotein genes15,16). However definitive conclusions have not been obtained and this finding suggests that these loci are not major causes to the disease.
Genetic studies of families with early-onset NIDDM have shown tight linkage with DNA polymorphisms in the adenosine deaminase gene on chromosome 2017) and with DNA polymorphisms in the glucokinase gene on chromosome 718,19). Although the diabetes-susceptibility gene on chromosome 20 has not been identified, the glucokinase gene has been considered a prime candidate for the diabetes-susceptibility gene on chromosome 73). Recently, Froguel et al20) also reported that glucokinase gene was tightly linked to the gene responsible for early-onset NIDDM. Glucokinase is the key component in glucose sensing by pancreatic islet β-cells and diabetes due to glucokinase mutations may be one of the most common single-gene disorders described to date.
In our RFLP study of glucokinase gene, 1.3 kb fragment was not found in 10% of NIDDM patients. These results suggest that the NIDDM patients of negative 1.3 kb allele of glucokinase gene may have the defect of hepatic glucokinase gene. thus hepatic glucokinase gene may abnormally express quantitatively or qualitatively. Hepatic glucose uptake and metabolism decrease, thus causing increased plasma glucose level. Increased plasma glucose would induce beta cell glucokinase expression followed by further increased insulin secretion. Despite the increased plasma insulin concentration, hepatic glucokinase would not be induced because of its defect. Plasma glucose concentration would be further increased. NIDDM patients without 1.3 kb allele/Pvu II digestion of glucokinase gene were characterized by increased insulin secretion compared to those NIDDM with 1.3 kb allele. Sequencing analysis and further characterization of 1.3 kb fragment should be followed to give us answers for the pathophysiologic importance of 1.3 kb allele.
Recently, Tanizawa et al21) revealed that the human glucokinase gene is composed of 12 exons that span a region of >50,000 base pairs of chromosome 7, band p13. This gene is transcribed from two tissue-specific promoters, one of which is active in the B cell and the other in hepatocyte. This feature allows the gene to be independently regulated in these two tissues. β-cell glucokinase mRNA is the product of exon 1a and exons 2–10, wereas the major liver glucokinase mRNA is derived from exon 1b and exons 2–10. The sequence information for glucokinase gene made it possible to clarify the molecular basis of potential glucokinase defects in NIDDM patients and may further elucidate the nature of genetic susceptibility to NIDDM.
Several different types of RFLPs have been described. In addition to nucleotide sequence substitutions, that change the cleavage site for a restriction endonuclease and the vairable number of tandem repeats, VNTR type DNA polymorphism and Alu variable poly (A), DNA polymorphisms have been characterized. In the human genome, interspersed repetitive element (dC-dA) n (dG-dT) n sequence has been shown to be abundent. These so-called (CA)n microsatellite repeat elements often exhibit size polymorphisms which are readily detected by PCR-based assays22,23). Tanizawa et al21) have previously identified a (CA)n microsatellite repeat about 8 kb downstream of the human glucokinase gene, which was highly polymorphic.
Population association studies are based on the assumption that a mutation causing a disease and a genetic marker are in linkage disequilibrium or that the marker is the disease locus24).
In our study with PCR-SSCP assay for 8 kb downstream microsatellite region, three different alleles were observed including Z, the most common allele, Z+2 and Z+4, No significant allele associated with NIDDM was noted. The most common genotype was Z/Z, which was more common in nondiabetic control than in NIDDM patients. In similar studies performed in many racial groups, Chiu et al25) reported that the Z allele was more common in American blacks with nondiabetic subjects than in diabetic patients and Z+4 allele was more common in diabetic patients. Noda et al26) also reported that the Z+4 allele was found more frequently in Japanese with diabetic than in nondiabetic subjects and −2 allele, 5′-flanking region, was common in diabetic than in nondiabetic subjects. Because some of the control subjects might develop diabetes later in life, this would be a factor that weighted against finding a difference in glucokinse allele frequencies between the two groups. In a study with Mauritian Creoles27), Z+2 was reported as an important risk factor for NIDDM, but not in Mauritian Indians. In our study, frequencies of Z+2 and Z+4 alleles were not significantly different in both groups. These racial discrepancies may represent the genetic heterogeneities of NIDDM in different racial groups and further studies are required to determine the prevalence of glucokinse mutations among subjects with NIDDM and to define the nature of this defect.
Glucokinase is major enzyme that phosphorylates glucose upon entry into liver and islet β-cell, Therefore, decreased cellular levels of glucokinase activity, as a consequence of the missense, nonsense and splicing mutations, may increase the glucose threshold for insulin secretion by the β-cell, as well as impair glucose uptake and metabolism by the liver. It has been suggested that a modest 15% decrease in glucokinase activity may shift the set point for glucose-induced insulin secretion from 5 to 6 mM4,28). Clinical studies of subjects with glucokinase mutations are consistent with a decreased sensitivity of the β-cell to glucose29). There may also be a correlation between the effect of the mutation on glucokinase activity and the severity of the glucose intolerance20). Mutations in glucokinase result in a form of NIDDM that has a very early age on onset, often in childhood. Moreover, NIDDM due to glucokinase mutations appears in general to be relatively mild, and many subjects with such mutations do not have overt NIDDM. In diabetics with positive polymorphic microsatellite repeat markers, Z+4 and −2 allele, no differences were found in the age of diagnosis, positive family, mode of therapy, current HbA1c or daily C-peptide immunoreactivity excretion compared to diabetics without those markers26). In this study, there was also no difference in clinical findings according to 3′ microsatellite genotypes in NIDDM.
In conclusion, NIDDM patients, without 1.3 kb allele/Pvu II digestion of glucokinase gene, were characterized by increased insulin secretion. The genotype of homozygote Z/Z was more common in normal subjects than in NIDDM patients. These data suggested that there does not appear to be a significant glucokinase allele associated with NIDDM but Z/Z homozygote genotype may play a suppressive role in the pathogenesis of a certain type of NIDDM in Korean. Although one should be cautious about the potential spurious observations in association studies, the significance of the results should also be recognized in the genetic analysis of heterogenous disorders like NIDDM. Further studies may be required to identify the molecular basis of this association.
Notes
This research was supported by grants from the Korea Science and Engineering Foundation (91-07-00-16) and the Korean Diabetes Association.