Autosomal Dominant Polycystic Kidney Disease: 2009 Update for Internists
Article information
Abstract
Because autosomal dominant polycystic kidney disease (ADPKD) is one of the most common genetic abnormalities seen in today's medical practice, many internists will likely treat patients affected by this condition. Genetic abnormalities have been increasingly recognized, and the pathophysiology of the disease is beginning to be unraveled. Because of advances in imaging technology, surrogate markers for disease progression have allowed clinical studies of newer therapeutic agents to proceed. In the near future, therapies for this common genetic disease may be available to either prevent or stabilize the disease course for many affected individuals.
Genetics
The frequency of ADPKD is estimated to be between 1 in 500 and 1 in 1,000 life births [1]. Two phenotypically similar forms of ADPKD are associated with mutations on chromosomes 16 (PKD1) and 4 (PKD2). Mutations in these genes encoding the proteins polycystin-1 and -2 lead to abnormalities in cell proliferation, apoptosis, tubular basement membranes, and tubular fluid secretion, ultimately resulting in slowly expanding renal cysts (Fig. 1). This disease is autosomal dominant with virtually complete penetrance of the abnormal genes. However, recent information suggests that some patients with milder disease may have incom-plete penetrance of the abnormal mutated gene [2]. A great deal of individual overlap exists in the phenotype between patients with the PKD1 mutation and the PKD2 mutation. As a group, however, patients with the PKD1 mutation progress more rapidly to end-stage renal disease and have a more severe renal insult compared to those with PKD2 [3], perhaps because patients with PKD2 form fewer cysts [4].
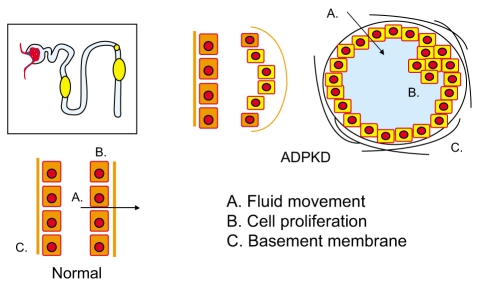
Tubular cell abnormalities in autosomal dominant polycystic kidney disease. Mutations on chromosome 16 and 4 encoding polycystin-1 and -2 lead to abnormalities in cell proliferation, apoptosis, tubular basement membranes, and tubular fluid secretion, ultimately resulting in slowly expanding renal cysts.
ADPKD is thought to be an example of a "second hit" phenomenon. An individual inherits a mutated polycystic kidney disease gene from one of his or her parents. The wild-type or normal allele in individual cells may then be affected by a so-called "second hit" allowing this cell to proliferate and form individual cysts [5]. No more than 5% of nephrons are thought to be involved in cystic change, but the genetic mutation is obviously present in all cells. Some manifestations of ADPKD, such as intracranial aneurysms, also persist in families.
Pathophysiology and pathobiology
The polycystin proteins are situated within the cell in areas involving the primary cilium. Thus, mutated polycystins are an example of a ciliopathy. The cilia's function is to transmit mechanosensory signals into the cell controlling cell growth and proliferation. When the polycystins are mutated, a complex series of events ensues, leading to growth of individual cells and then to the formation of a fluid-filled cyst [6,7]. As the cyst gradually enlarges, it becomes pinched off from the nephron of origin. Because fluid secretion continues, the result is a gradually enlarging cystic kidney with an increased volume. The renal parenchyma may also show abnormalities, but the primary pathophysiologic process of the cystic kidney is a gradually enlarging kidney with an increase in renal volume. Recent data have suggested that the progression of renal disease, the occurrence of hypertension, and symptoms of the disease are related to kidney volume [8,9]. In fact, in most cases, renal volume increases years before the glomerular filtration rate starts to decrease, making the glomerular filtration rate or its surrogate serum creatinine a poor way to follow patients with ADPKD.
Diagnosis
An ADPKD diagnosis is usually made by renal imaging, and ultrasound is most frequently used. Ultrasound criteria for diagnosis have been recently published, based on ultrasound examinations with known ADPKD type 1 and type 2 compared to the ultrasound frequency of cysts in a normal unaffected population [10]. Thus, the number of cysts correlated with patient age helps make the diagnosis. A single negative ultrasound examination in a young person at risk for ADPKD because of family history does not necessarily exclude the diagnosis. While magnetic resonance imaging (MRI) and computed tomography scanning are perhaps slightly more sensitive for detecting cysts, the cost of these procedures is much greater than ultrasonic examination and less widely available. At present, ultrasound should be used preferentially for diagnosis. With modern advances in genetics, the DNA sequence of individual patients can be determined. However, these expensive tests are not widely utilized at the present time, except in unusual cases to confirm a questionable diagnosis or to exclude a possible organ donor in a patient with a family history of ADPKD [11].
Course and prognosis
Eventually, with ADPKD, all patients develop progressive involvement of the kidneys. Each patient has his or her signature rate of cyst growth [8]. In general, patients with PKD-1 reach end-stage renal disease about 10-15 years earlier than those with the PKD2 mutation. Note that not all patients who carry this genetic abnormality reach end-stage renal disease and many patients, particularly those with type 2 disease, die prior to chronic kidney disease becoming a clinical problem. The renal volume of a patient as determined by MRI is an important prognostic marker in an individual patient regarding the likelihood to progress. These changes in renal volume have been detected by sophisticated MRI studies, and a population of patients with slower progression and another group with faster progression appear to exist [8,9]. All of the risk factors for progression have not been determined, but a large renal volume is associated with a decrease in renal blood flow as indicated by MRI studies, hypertension, and male gender [9].
Signs and symptoms
Many patients with ADPKD are completely asymptomatic. A minority of patients will have early clinical symptomatology such as urinary tract infections, gross hematuria, or abdominal pain, which brings them to medical attention. At that point, imaging studies will confirm the diagnosis. However, the vast majority of patients have few signs and symptoms and often are diagnosed because of their positive family history or the development of hypertension. Hypertension is probably the most remediable and serious complication of ADPKD [12]. The new onset of hypertension in a patient at risk for polycystic kidney disease should prompt aggressive treatment and diagnostic studies. Patients with hypertension during the course of ADPKD have early and more severe left ventricular hypertrophy. The pathogenesis of hypertension in ADPKD is thought to be due to activation of the renin-angiotensin system (Fig. 2). Thus, medications that interfere with that system, such as angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, are the preferred form of therapy. However, the evidence that these drugs are better than other drugs that control hypertension to a similar degree are not robust and the choice of antihypertensive therapy is largely based on theoretical considerations [13]. Studies examining the efficacy and the degree to which blood pressure should be lowered in ADPKD are now under way.
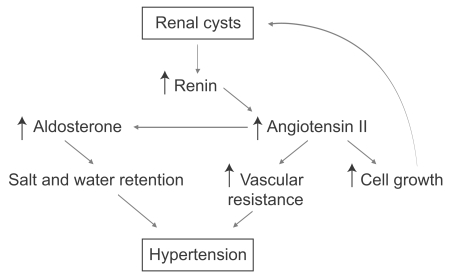
Mechanisms of hypertension in autosomal dominant polycystic kidney disease (ADPKD). The pathogenesis of hypertension in ADPKD is thought to be due to activation of the renin-angiotensin system.
Other symptoms of ADPKD include the occurrence of abdominal, back, and flank pain. This pain can be severe in many individuals and require pain management. Nonsteroidal anti-inflammatory agents have renal effects particularly in patients with an activated reninangiotensin system and thus are to be avoided. This leaves individuals with persistent chronic abdominal pain in a therapeutic dilemma. These patients often benefit from a careful approach to their symptoms. Pain adjuvants such as antidepressants and muscle relaxants, and in rare cases, interventions designed to reduce cyst volume surgically or laparoscopically, are often needed. Gross hematuria and acute abdominal pain may be secondary to rupture of individual cysts. Usually these are managed conservatively but in recurrent cases, interventional radiologic procedures are necessary [14].
Urinary tract infections during the course of ADPKD are common. When the renal parenchyma and cysts are involved, these patients can be refractory to the usual antibiotics necessary to treat urinary tract infections. Some individual antibiotics are known to penetrate cysts well, whereas other common antibiotics do not appear in cyst fluid and urine in sufficient concentrations to eradicate a parenchymal infection. Antibiotic groups that are preferred in patients with parenchymal involvement in ADPKD include trimethoprim-sulfamethoxazole, floroquinolones, and chloramphenocol [15,16]. Note that most patients with ADPKD and urinary tract infections have the usual types of upper and lower urinary tract infections that can be managed with many antibiotic agents. Only those infections involving the parenchyma, which are refractory to therapy, require cyst-penetrating antibiotics. The possibility exists for patients with parenchymal infections to be febrile and have a negative urine culture because not all cysts are in continuity with the urinary space. A patient with prolonged fever, failure to thrive, weight loss, and nonspecific gastrointestinal symptoms should be evaluated for an occult renal parenchymal infection. Such infections can be localized with various radiographic procedures but remain a difficult therapy problem.
Extrarenal manifestation of ADPKD
The most common extrarenal manifestation of ADPKD is hepatic cystic involvement. Quite often these cysts are incidental findings and not clinically significant, although in women, these cysts can enlarge rapidly particularly under the influence of multiple pregnancies and hormonal therapy. They then can become the dominant clinical problem. Although hepatic failure is uncommon, symptoms related to a massively enlarged cystic liver include loss of appetite, weight loss, esophageal reflux, and abdominal discomfort, which interfere with the activities of daily living. Under these circumstances, specialized care for reducing hepatic cyst volume may be required to relieve symptoms. In rare cases, the cystic liver dominates the clinical picture and renal involvement is less important.
Intracranial aneurysms are perhaps the most severe extrarenal manifestation of ADPKD, although aneurysms are present only in approximately 5% of all patients with ADPKD. Those with a family history of early death due to stroke or prior ruptured intracranial aneurysms are much more likely to have these aneurysms. Ruptured aneurysms depend on the size attained, and most ruptures occur in aneurysms that are greater than 10 mm in size. Smaller aneurysms can be managed expectantly, but frequent surveillance every 1-2 years is probably wise. If an aneurysm attains a large size, even if patients are asymptomatic, intervention should be accomplished either with surgery or with radiographic techniques. The benefit of treatment is often counterbalanced by the complications of the procedure, but a ruptured intracranial aneurysm carries a 50% mortality rate.
Other extrarenal manifestations include a propensity to hernias, abdominal or inguinal, and a very high incidence of diverticulosis. A patient presenting with acute abdominal pain poses a complex problem for differential diagnosis in that ruptured cysts, cyst infection, and acute diverticulitis can look remarkably similar clinically. Cardiac valvular disease of the mitral valve and tricuspid valves, namely prolapses, are more frequent in patients with ADPKD, but these seldom lead to clinical problems [17,18].
Therapies
A patient with ADPKD managed by internists should have careful blood pressure control and control of other cardiovascular risk factors such as tobacco smoking, hyperlipidemia, body weight management, and aggressive control of diabetes if present. Based on natural history studies involving MRI determinations of annual increases in cyst volume, therapeutic trials of agents shown to be beneficial in animal models of cystic disease are anxiously awaited. Two such agents are the vasopressin 2 receptor antagonist, tolvaptan, and the mTOR inhibitor, sirolimus. These drugs interfere with cyst growth in experimental animal models, and early data suggest they are well tolerated in humans. Stabilization or a decrease in cyst volume will be the outcome measure used to gauge therapeutic efficacy in these ongoing clinical trials [19]. Other drugs that interfere with cyst growth pathways exist, which are being elucidated in basic science laboratories and are candidates for therapeutic intervention. If these therapies prove useful, screening of individuals at risk for ADPKD will become more important because early initiation of such therapy may prevent the progression to chronic kidney disease or even end-stage renal failure.