Comparison of the Clinical Manifestations, Brain MRI and Prognosis between NeuroBehçet's Disease and Neuropsychiatric Lupus
Article information
Abstract
Background
Neuropsychiatric systemic lupus erythematosus (NPSLE) shows some similarities to neuroBehçet's disease (NBD) in that both conditions have some analogous clinical features and they are both pathologically associated cerebral vasculopathy. This study compared the clinical manifestations, brain MRI findings and prognosis of NPSLE and NBD patients.
Methods
Forty three patients with NPSLE (n = 25) or NBD (n = 18), who were monitored at a single center, were enrolled in this study. We retrospectively analyzed the clinical and brain MRI data. The neuropsychiatric manifestations were classified in both groups according to the new American College of Rheumatology nomenclature for NPSLE.
Results
The diffuse symptoms that included mood disorders, psychosis, confusion, cognitive dysfunctions, generalized seizures and headaches other than migraine or cluster headaches were more commonly observed in the NPSLE patients, while the frequency of focal diseases such as cranial neuropathy tended to be higher in the NBD patients. The brain MRI revealed that the NBD patients had more abnormalities in the brain stem than did the NPSLE patients. Most of the patients improved, at least partially, after being treated with glucocorticoid and/or immune suppressants. However, the disease course differed significantly between the two groups. There were more episodic cases in the NPSLE group of patients, while there were more remittent cases in the NBD group of patients.
Conclusion
NPSLE had a tendency to cause diffuse neuropsychiatric manifestations, and it has a different predilection of brain lesions compared with NBD. The NBD patients showed a poorer outcome than did the NPSLE patients, suggesting that different therapeutic strategies for the two diseases need to be considered.
INTRODUCTION
Behçet's disease is a chronic inflammatory disease of an unknown origin, and it is characterized by the triad of oral ulcerations, genital ulcerations and uveitis. A number of additional features are commonly observed: these include arthritis, retinal and cutaneous vasculitis, deep vein thrombosis, gastroenteric disorders and neuropsychiatric involvement1). Among these features, neuropsychiatric involvement is one of the most serious complications. The reported frequency of neuroBehçet's disease (NBD) in Behçet's disease patients ranges from 2.2% to 49%; however, the previous reports that employed large subject populations have reported a rate of approximately 5%2, 3). The spectrum of NBD varies and this can be classified into two major forms. One form is attributable to small venous inflammatory disease with focal or multifocal central nervous system (CNS) parenchymal involvement, and this form is seen in the majority of patients. The other form is caused by a cerebral venous sinus thrombosis with limited symptoms, and this form has a better neurological prognosis4). It is quite rare for the two types of involvement to occur simultaneously in the same individual2, 3). In addition, psychiatric symptoms and peripheral nervous system (PNS) involvement is rarely encountered4).
Systemic lupus erythematosus (SLE) is an autoimmune inflammatory disease that might lead to multi-organ failure with various clinical courses and a variable prognosis, and SLE is tightly linked to the overproduction of a range of autoantibodies5). Neuropsychiatric SLE (NPSLE), as well as lupus nephritis, is a major cause of morbidity and mortality in SLE patients6). The prevalence of NPSLE ranges from 20% to 60% of SLE patients, whch most likely depends on the different inclusion criteria used in the studies7). The spectrum of NPSLE also varies, and this includes neurologic syndromes of the central, peripheral and autonomic nervous system. Psychiatric syndromes are also commonly observed in the SLE patients for whom other causes have been excluded8).
In addition to the clinical diversity, the difficulty in defining the outcome measures and the lack of a standard terminology are considered obstacles for understanding the disease mechanisms and for evaluating the level of therapeutic intervention in both NBD and NPSLE patients. The American College of Rheumatology (ACR) has recently formulated specific criteria for NPSLE. The new nomenclature includes case definitions, reporting standards and the diagnostic testing recommendations for 19 different neuropsychiatric syndromes8). These definitions may provide a reliable classification for recruiting homogenous groups of patients for designing randomized controlled trials6).
NPSLE has some similarities to NBD in that both conditions have some common clinical features and they are pathologically associated with cerebral vasculopathy. Also, in Korea, SLE and Behçet's disease are the representative autoimmune diseases involving the nervous system. Therefore, this study compared the clinical characteristics, brain MRI findings and prognosis between NPSLE and NBD patients at a single center. We demonstrate here that NPSLE has a propensity to cause diffuse neuropsychiatric manifestations, and it shows different predilections for brain lesions compared with NBD. Moreover, the poorer outcome in NBD compared with NPSLE suggests the need for different treatments strategies for the two diseases.
MATERIALS AND METHODS
Patients
This study included 43 patients with NPSLE or NBD and these patients were monitored at Kangnam St. Mary's hospital between January 1999 and December 2002. The patients were identified from the hospital's automated information system. The patients' information was analyzed by retrospectively reviewing the data registered at regular intervals for at least 3 months. Of the patients who were screened, 25 NPSLE patients fulfilled the 1982 Revised Criteria for SLE as set forth by the American Rheumatism Association9), and 18 NBD patients met the 1990 criteria of the International Study Group for Behçet's disease10). Three of NPSLE patients had antiphospholipid syndrome that fulfilled both the laboratory and clinical Sapporo preliminary criteria11). A written informed consent was obtained from each subject after giving a complete explanation of the study to the subjects. Three NPSLE patients and 3 NBD patients were referred to this center from local clinics as a result of their uncontrolled symptoms, and their medical records from those clinics were thoroughly reviewed.
Data collection and analysis
The clinical data was comprised of gender, the patient's age at the time of the neuropsychiatric event, the disease duration prior to the neuropsychiatric attack, the follow-up duration, the neuropsychiatric manifestations, the type and dosage of drugs and the clinical course. A team of rheumatologists, neurological and psychiatric consultants evaluated the level of neuropsychiatric involvement. Those patients with secondary neuropsychiatric events that were not directly related to SLE were excluded. The neuropsychiatric manifestations were classified according to the 1999 ACR Nomenclature and Case Definitions for neuropsychiatric lupus syndromes8), and all the enrolled patients met these criteria. Using this classification, the presentations were subdivided into one of the following groups; diffuse disease (including psychosis, mood disorder, cognitive dysfunction, acute confusional states, headaches other than migraine or cluster headache, and generalized seizures), focal disease (migraine or cluster headache, focal seizures and other neuropsychiatric manifestations) and complex presentations (cases where both diffuse and focal neuropsychiatric manifestations occurred simultaneously). For making comparison with NPSLE, the neuropsychiatric manifestations of NBD were also classified with using the same nomenclature used for NPSLE. All the NBD patients met these criteria. In particular, regarding headache, the patients who responded to conventional medication and had other explainable causes were excluded due to some discrepancy in the relationship between headache and NPSLE or NBD.
As the part of the initial evaluation, all the patients underwent routine laboratory tests, chest radiographs and electrocardiograms. The sera of the NPSLE patients were tested for antinuclear antibodies, the C3 and C4 complement components, antibodies to extractable nuclear antigens, antibodies to double-stranded DNA as assessed by radioimmunoassay, and anticardiolipin antibodies (ACA) and anti-β2-glycoprotein I antibodies (anti β2-GPI antibodies) as assessed by ELISA. Positive levels of ACA and anti β2-GPI antibodies were defined as being levels > 5 standard deviations above the mean levels of 100 healthy controls. Where indicated, some of the patients underwent cerebrospinal fluid analysis, electroencephalograms and echocardiograms.
Each patient was treated differently, according to their disease severity and possible causes. Treatment was classified as 1) conservative therapy, which was composed of neurological or psychiatric regimens, and 2) immune suppressive therapy with high dose oral prednisolone (1-2 mg/kg/day) and/or other immunosuppressive agents that included cyclophosphamide, azathioprine, chlorambucil, methotrexate, cyclosporine, interferon-α (IFN-α) and anti-tumor necrosis factor-α (anti-TNF-α). The treatment response and clinical course, that is, episodic (a single episode of neuropsychiatric manifestations), remittent (recurrent episodes of remission and relapse of neuropsychiatric manifestations), and sustained or progressive (persistent neuropsychiatric manifestations without remission), were also analyzed in each patient. The treatment response was classified into a good, partial and poor response. A good response was defined as the complete improvement of neuropsychiatric symptoms without any sequela. A partial response was defined as the initial improvement with later exacerbation and/or incomplete improvement with sequela. A poor response was defined as no improvement and/or exacerbations.
Brain magnetic resonance imaging (MRI)
All the patients underwent brain MRI to determine the causes of their neuropsychiatric symptoms, and the MRI included T1-wighted images, T2-weighted images and fluid-attenuated inversion-recovery (FLAIR) images. The MRI was performed using a 1.5 T MRI system (GE SIGNA Advantage version 4.8). The conventional spin echo pulse sequence with a TE of 20 ms and a TR of 400 ms was used to obtain the T1-weighted MR images. The fast spin echo pulse sequence with a TE of 90 ms and a TR of 2,500 ms was used to obtain the T2-weighted MR images. Contrast medium was administered to all patients, and this was followed by obtaining the axial/sagittal/coronal T1-weighted images. Two neuroradiologists, who were unaware of the clinical data, evaluated the MRI. For analysis, the abnormal MRI findings were carefully classified into the following categories: an abnormal T2 weighted image, infarct-like lesions (moderate-size to large, roughly wedge-shaped areas of abnormal high signal on the T2-weighted images and/or encephalomalacia involving the gray and white matter), parenchymal hemorrhage, loss of brain volume, abnormal intracranial enhancement and meningeal enhancement. The location of the brain lesions was also analyzed. The lesions were classified as stable, resolving or progressing if the patients underwent MRI more than once.
Statistical analysis
Comparisons of the numerical data between the groups were performed using the Mann-Whitney rank sum test, and the categorical data was analyzed using chi-square tests or Fisher's exact probability tests. p values < 0.05 were considered to be significant.
RESULTS
Demographic and extra-neuropsychiatric features
The demographic characteristics of the 43 patients (25 NPSLE patients and 18 NPBS patients) are summarized in Table 1. The NPSLE patients showed a female predominance (p=0.001). The age at the time of the neuropsychiatric event was higher for the NBD patients (38±9 years) than for the NPSLE patients (31±10 years) (p=0.024). The disease duration prior to the neuropsychiatric attack and the length of the follow-up period was similar for the two groups. For the NPSLE, 3 patients (12.0%) had antiphospholipid syndrome, 7 patients (28.0%) had lupus nephritis, 2 patients (8.0%) had mesenteric vasculitis and 5 patients (20.0%) had hematological abnormalities that affected the treatment strategies. For the NBD, all the patients had recurrent oral ulcerations, 7 patients (38.9%) had genital ulcerations, 8 patients (44.4%) had eye lesions, 15 patients (83.3%) had skin lesions such as erythema nodosum and folliculitis, 1 patient had gastrointestinal involvement, 8 patients (44.4%) had arthritis and 1 patient had major arterial involvement.
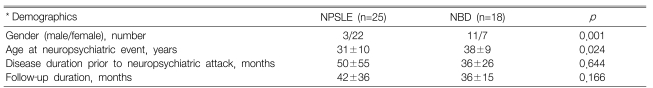
Demographic data for the neuropsychiatric SLE (NPSLE) and neuroBehçet's disease (NBD) patients
Neuropsychiatric manifestations
Table 2 shows the neuropsychiatric manifestations in the two groups. As initial manifestations, the neuropsychiatric complications were the major symptoms and signs in 5 SLE patients and 3 Behçet's disease patients. Four NPSLE patients and 4 NBD patients had a history of previous neuropsychiatric complications. These include mononeuritis multiplex, seizure and transverse myelitis for the NPSLE patients, and aseptic meningitis, cerebrovascular disease (CVD) and cognitive dysfunction for the NBD patients. Sixteen (64.0%) NPSLE patients showed evidence of concomitant extra-neuropsychiatric complications (e.g. lupus nephritis and mesenteric vasculitis) and/or elevated SLE activity, which was determined by the levels of C3, C4 and the anti-double-stranded DNA antibody.
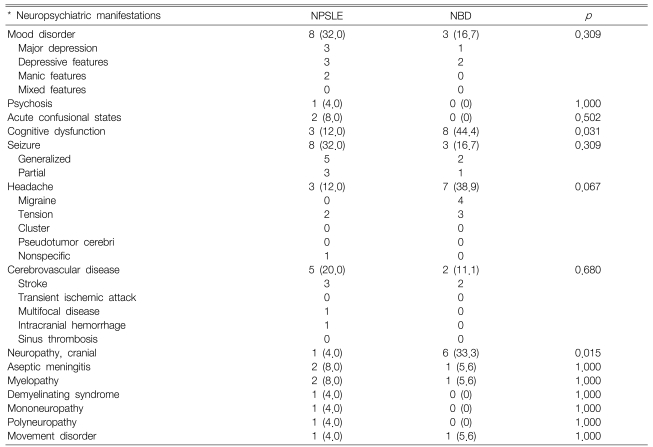
Comparison of the neuropsychiatric manifestations between the neuropsychiatric SLE (NPSLE) and neuroBehçet's disease (NBD) patients according to the ACR case definitions for NPSLE
The common features in the NPSLE patients were mood disorders (32.0%), seizure (32.0%), and CVD (20.0%). In contrast, the common features in the NBD patients were cognitive dysfunction (44.4%), headache (38.9%), and cranial neuropathy (33.3%). Cognitive dysfunction and cranial neuropathy were more common in the NBD patients. All the patients with cognitive dysfunction in both groups were associated with a variety of neurologic or psychiatric diseases, including neuropathy, headache, CVD, movement disorder and mood disorders. Half of the cranial neuropathy of the NBD patients (3 of 6) was associated with cognitive dysfunction. The types of cranial neuropathy of the NBD patients were vestibulecochlear dysfunction (4 of 6 patients) and oculomotor dysfunction (2 of 6 patients). One NPSLE patient had optic nerve dysfunction.
Importantly, Table 3 showed the subgroup analysis, and this revealed that diffuse manifestations were more common in the NPSLE patients (56.0 versus 16.7%, respectively, p=0.013). The frequency of focal manifestations and complex manifestations was similar for both groups.

Subgroup analysis of the neuropsychiatric manifestations
All the NPSLE patients underwent tests for the ACA and/or anti β2-GPI antibodies. A minimum of 2 positive tests (either ACA and/or anti β2-GPI antibodies) were required before the patient was confirmed to be positive for anti-phospholipid antibody. The result showed that 13 NPSLE patients (52.0%) had increased levels of the anti-phospholipid antibody with a high titer (> 5 SD over the mean levels of the healthy controls); 7 patients were positive for ACA and 13 patients tested positive for anti β2-GPI antibodies, respectively. Three of 8 patients with seizure disorder had increased levels of ACA and anti β2-GPI antibodies. In addition, 2 of 5 patients with CVD and all 3 patients with cognitive dysfunction tested positive for antiphospholipid antibody. The other neuropsychiatric features that were associated with a positive test for anti-phospholipid antibody included mood disorders, acute confusional states, psychosis, transverse myelitis, cranial neuropathy and movement disorders.
MRI findings
As shown in Table 4, the NBD and NPSLE patients had different predilections of brain lesions. For example, the brain stem lesions on MRI were more frequently encountered in the NBD patients (55.6 versus 20.2%, respectively, p=0.024), whereas the frequency of the cerebral white matter abnormalities tended to be higher in the NPSLE patients than in the NBD patents (44.0 versus 22.2%, respectively) (Table 4, Figure 1). Abnormalities in the cerebral white matter of the NPSLE patients were mainly located in the frontal and parietal lobes.
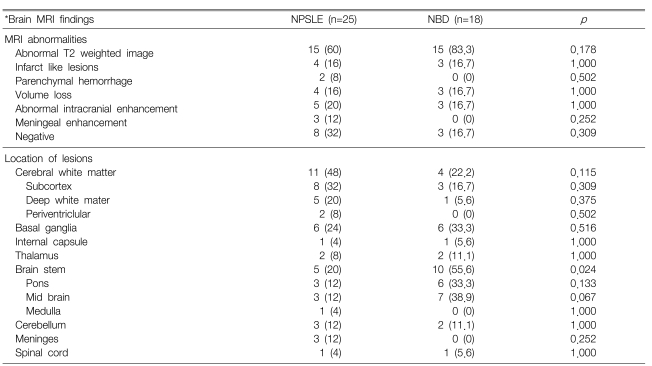
Comparison of the brain MRI findings in the neuropsychiatric SLE (NPSLE) patients and the neuroBehçet's disease (NBD) patients

Representative MRI findings in a neuroBehçet's disease (NBD) patient and a neuropsychiatric SLE (NPSLE) patient. (A) Fluid-attenuated inversionrecovery (FLAIR) images of a 43-year-old male NBD patient with cranial neuropathy. There are abnormal signals in the brain stem and the left frontal white matter. (B) FLAIR images of a 15-year-old female NPSLE patient with generalized seizure. There are abnormal signals in the subcortical and periventricular white matter.
It's interesting that 8 (32.0%) of the 25 NPSLE patients and 3 (16.7%) of the 18 NBD patients had a normal brain MRI, despite having definite neuropsychiatric symptoms and signs. Of these, 5 (62.5%) of the 8 NPSLE patients had diffuse neuropsychiatric manifestations as their major symptom, whereas all 3 NBD patients presented with focal manifestations such as seizure and movement disorders. Parenchymal volume loss was observed in 4 NPSLE and 3 NBD patients. An abnormal high signal was typically high on the FLAIR and other T2-weighted images, and this was observed in 15 (60.0%) NPSLE patients and 15 (83.3%) NBD patients. However, this was frequently nonspecific, and this was interpreted as being consistent with focal ischemia, demyelination or vasculitis. Infarct-like lesions were observed in 4 NPSLE patients and 3 NBD patients. Parenchymal hemorrhage was observed in only 2 NPSLE patients. Five NPSLE patients and 3 NBD patients showed abnormal intracranial enhancement, mainly with focal and heterogonous distributions. Other minor abnormalities included meningeal contrast enhancement in the NPSLE patients, and internal carotid artery narrowing, venous angioma, lipoma and mega cisterna magnata in the NBD patients.
The follow-up MRI, taken at an interval of from 1 month to 2 years, was available for 7 NPSLE patients and 3 NBD patients. For the NPSLE patients, two showed complete regression, two showed partial regression, two showed progression and one showed no change. For the NBD patients, two showed regression and one did not show any change on the follow-up MRI.
Therapy and clinical course
Table 5 and Figure 2 show the therapy, treatment response and clinical course of the patients. The maintenance therapy of all the patients included prednisolone at the time of the attack for treating a variety of causes. The percentage of patients receiving conservative or immune suppressive therapy was not significantly different between the NPSLE patients and NBD patients (e.g. conservative treatment: 32.0% for the NPSLE patients and 22.2% for the NBD patients). After the neuropsychiatric attack, 16 NPSLE patients (64%) and 7 NBD patients (38.9%) were given high dose oral prednisolone therapy (1-2 mg/kg/day) (p=0.766). Among them, 8 NPSLE patients (32%) and 5 NBD patients (27.8%) were given intravenous methylprednisolone pulse therapy (500 mg-1g daily for three days) (p=0.153). The number and type of immune suppressants (e.g. cyclophosphamide, azathioprine) that were newly added and then maintained after the neuropsychiatric attack was similar in the two groups with cyclosporine being the only exception (Table 5). Three NPSLE patients with antiphospholipid syndrome were also treated with an anticoagulant. Three NPSLE patients with intractable thrombocytopenia or thrombotic thrombocytopenic purpura were treated with high dose prednisolone plus intravenous immunoglobulin and/or plasmapheresis. Two NBD patients (one with CVD and one with cranial neuropathy along with cognitive dysfunction) had progressive disease despite being treated with high dose prednisolone plus immune suppressants. Therefore, biological agents such as IFN-α and anti-TNF-α monoclonal antibody were added to the above-mentioned immune suppressants for treating these 2 patients.
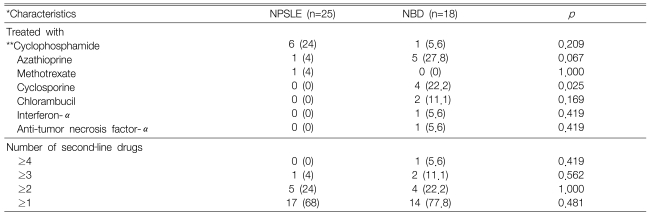
Comparison of the immune suppressants newly added and maintained after neuropsychiatric attack in the neuropsychiatric SLE (NPSLE) and neuroBehçet's disease (NBD) patients
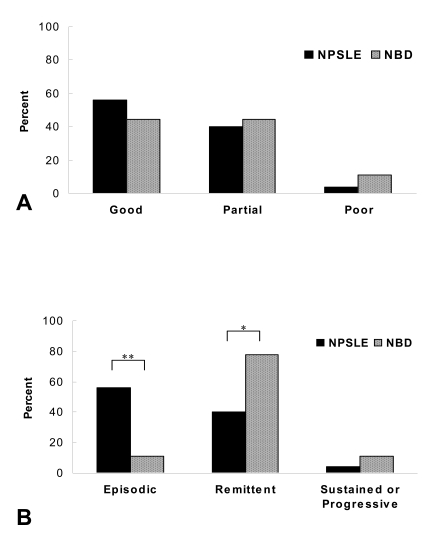
Comparison of the initial treatment response (A) and disease course (B) after conservative or immune suppressive treatment for neuropsychiatric SLE (NPSLE) and neuroBehçet's disease (NBD) patients. The data is presented as a percentage. *, p < 0.05; **, p < 0.01.
For most patients, the neuropsychiatric manifestations improved after being treated with conservative or immune suppressive treatments (88.0% for the NPSLE patients and 88.9% for the NBD patients) (Figure 2A). However, the disease course was significantly different between the two groups after the initial improvement. Episodic cases were more common in the NPSLE patients (65.0 versus 22.2%, respectively, p=0.012), while remittent cases were more frequently noted in the NBD patients (72.2 versus 24.0%, respectively, p=0.02) (Figure 2B). Two NBD patients showed a good or partial response with the additional IFN-α or anti-TNF-α monoclonal antibody treatment. However, the biological agents failed to prevent further attacks or progression of disease. The number of patients who showed a sustained or progressive course that was refractory to therapy was similar in the two groups.
DISCUSSION
Systemic immune-mediated diseases can often affect the nervous system12, 13), and among them, SLE and Behçet's disease are the common autoimmune diseases of the nervous system in Korea. NPSLE is believed to have a particular propensity to cause diffuse manifestations or psychiatric disorders14). In contrast, NBD is commonly associated with CNS parenchyma and central venous sinus thrombosis, and it rarely presents as psychiatric disorders4). However, no trial has been carried out to compare the two diseases, even though both conditions have been associated with immune-mediated mechanisms and they have pathologically revealed small vessel vasculopathy in the parenchyma of the nervous system. The classic histopathological studies of the nervous system in SLE patients have shown vascular changes and the resultant microinfarts, although any true vasculitis was rare14). As for NBD, vasculitis is believed to be the underlying pathological process of Behçet's disease and the pattern of the parenchymal lesions seen on cranial MRI was consistent with small vessel vasculitis and inflammation of veins. However, there are contradictory reports on the observed pathology and the observed histopathologic changes cover a wide spectrum, including vascultis, low grade inflammation, demyelination and degenerative changes. Definite vasculitis has not been observed in all cases13). Our current study was performed in a single hospital, and it showed that the NPSLE patients had a female predominance, and they were generally younger than the NBD patients. In addition, the NPSLE patients had a greater propensity for diffuse manifestations than the NBD patients, even though the possibility of a referral bias cannot be completely ruled out. There is a serious doubt regarding the previously suggested relationship between headache and SLE7), whereas headache is considered the most common neurological symptom observed in Behçet's disease patients4). In contrast, the frequency of focal diseases such as cranial neuropathy or headache tended to be higher in the NBD patients. These findings show a different pattern of neuropsychiatric presentations between the NPSLE and NBD patients, and they suggest that NPSLE might be caused by a different immunologic process from NBD.
The main problem is how to plausibly explain this discrepancy. The spectrum of the syndromes seen in NPSLE and NBD patients probably reflects multiple pathological processes, but ischemia is widely accepted to play a major role in causing CNS disorders in SLE patients, and reperfusion of an ischemic area carries the risk of edema and hemorrhage15, 16). The wide range of antibodies is a distinctive immunological feature of SLE, which is in contrast to Behçet's disease. These autoantibodies are believed to play two different, but not mutually exclusive roles in mediating neurological injury; direct injury to the neural target cells and antibody-induced rheological disturbances leading to an infarction14). Of the two mechanisms, antibody-mediated diffuse neural damage might be a possible mechanism for the propensity of NPSLE patients to exhibit diffuse manifestations, while antibody-induced rheological disturbances are widely accepted to be the major event for causing CVD. In this study, the high prevalence (52%) of anti-phospholipid antibodies with a high titer in the NPSLE patients supports this hypothesis.
MRI is a useful tool for detecting CNS lesions in patients suffering with SLE and Behçet's disease17, 18). Up to 70% of NPSLE patients have multiple white matter lesions seen on brain MRI18). The lesions appear on the T2 images as high signal areas in the periventricular and subcortical white matter, and they usually appear as normal in the T1-weighted images17). Neuro-imaging studies on NBD patients have shown that these lesions are generally located within the brain stem, occasionally with an extension to the diencephalons, or less commonly within the periventricular and subcortical white matter4, 18). These lesions are generally observed as high signal intensity on the T2-weighed images18). This current study also showed different predilections of MRI involvement in the NPSLE patients versus the NBD patients, which is consistent with earlier reports4, 17-19). The lesion predilection for the brain stem together with the sparseness of cortical lesions might explain the rarity or lack of higher cortical function disturbances in the NBD patients. The reason for the lesion predilection in the brain stem, basal ganglia and diencephalons is not known. It has been hypothesized that inflammation of the small-to-medium sized veins of those regions might be responsible for causing venous infarcts20). Although this appears to be reasonable, more pathological studies are needed to support this hypothesis.
In this study, 8 (32%) of the 25 NPSLE patients and 3 (16.7%) of the 18 NBD patients had a normal MRI, despite having definite neuropsychiatric complications. Of note, 5 of the 8 NPSLE patients with a normal MRI had diffuse manifestations. A neuroneolytic effect of the antibodies in SLE patients would be difficult to reconcile with the transient nature of many of the neurological features, as well as with the absence of clear pathological changes of the selective neuronal cell loss. One possibility would be the presence of non-lethal autoantibodies in the SLE patients, which could elicit a transient alteration of the cell function without causing cell death or inflammation. This non-lethal injury might present as an absence of specific MRI abnormalities in many SLE patients who have diffuse presentations. In parallel with this hypothesis, a more sensitive imaging tool such as 1H magnetic resonance spectroscopy is currently being introduced to detect the neuometabolite changes that precede permanent neuronal loss21).
The treatment for NPSLE and NBD should be adjusted according to the requirements of the individual patient because the neurological involvement in SLE and Behcet's disease is not uniform4, 22). Symptomatic, immunosuppressive and anticoagulant therapies are the main strategies for managing NPSLE and NBD. In this study, the neuropsychiatric manifestations were at least partially improved in almost all patients of both groups. However, the prognosis was significantly different between the patients of the two groups. The NBD patients, who showed a good response initially, they tended to be remittent, while the NPSLE patients tended to be episodic. This discrepancy suggests that NBD should be treated more aggressively in order to prevent its recurrence or progression. Unfortunately, there is little evidence on the efficacy of various treatment modalities for any form of neurological treatment in Behçet's disease4). Biological agents, including the IFN-α antibody and anti-TNF-α antibody, have recently been anecdotally reported to be of benefit for treating some of the systemic manifestations of Behçet's disease. However, none of these agents have been shown to be effective in a properly designed study4). In this study, the IFN-α or anti-TNF-α antibody failed to prevent further neurological attacks or progression in the two NBD patients.
This report is the first to describe the clinical findings, brain MRI and prognosis of NPSLE patients and NBD patients in a single hospital. The differences in neuropsychiatric presentations, the location of the brain lesions on MRI and the clinical course between the NPSLE patients and NBD patients suggest that there might be a different pathological process involved in each disease, and these 2 diseases should be treated differently in order to prevent recurrent attacks or progression of disease. However, this study was limited by its retrospective nature and the small number of subjects. Prospective multi-center trials are needed to provide more clues for understanding the disease's pathogenetic mechanisms, to guide further research on their cause and to find more effective treatments for these serious and potentially life-threatening diseases.
Notes
Supported by a grant from MOST (KOSEF) through The Systems Bio-Dynamics Research Center