Vitamin D and chronic kidney disease
Article information
Abstract
Chronic kidney disease (CKD) has been recognized as a significant global health problem because of the increased risk of total and cardiovascular morbidity and mortality. Vitamin D deficiency or insufficiency is common in patients with CKD, and serum levels of vitamin D appear to have an inverse correlation with kidney function. Growing evidence has indicated that vitamin D deficiency may contribute to deteriorating renal function, as well as increased morbidity and mortality in patients with CKD. Recent studies have suggested that treatment with active vitamin D or its analogues can ameliorate renal injury by reducing fibrosis, apoptosis, and inflammation in animal models; this treatment also decreases proteinuria and mortality in patients with CKD. These renoprotective effects of vitamin D treatment are far beyond its classical role in the maintenance of bone and mineral metabolism, in addition to its pleiotropic effects on extra-mineral metabolism. In this review, we discuss the altered metabolism of vitamin D in kidney disease, and the potential renoprotective mechanisms of vitamin D in experimental and clinical studies. In addition, issues regarding the effects of vitamin D treatment on clinical outcomes are discussed.
INTRODUCTION
Chronic kidney disease (CKD) is characterized by a progressive loss of renal function that often leads to end-stage renal disease (ESRD), high risk for cardiovascular disease, and high mortality [1,2]. CKD is a growing global health problem due to increased prevalence worldwide. In the United States, the prevalence of CKD was estimated to be 11% based on data from the Third National Health and Nutrition Examination Survey (NHANES III) [3]. In Korea, the prevalence of CKD stage 3 to 5 was 7.7% in 2007 based on data from the Korean National Health and Nutrition Examination Survey [4]. Under current guidelines, the therapeutic strategies for patients with CKD include aggressive blood pressure control, and medications such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and statins. However, despite these treatment options, individuals with CKD still have high rates of morbidity and mortality [1,5].
Over the years, studies have improved the understanding of the biological and clinical consequences of the interaction between disordered vitamin D metabolism and CKD. Vitamin D (e.g., 25-hydroxyvitamin D [25(OH)D3] and 1,25-dihydroxyvitamin D [1,25(OH)2D3]) deficiencies are now becoming a global epidemic problem in both the general population and patients with CKD [6,7]. Several observational studies have demonstrated an important link between vitamin D deficiency, impaired glomerular filtration rate (GFR), and increased mortality in patients with CKD [8,9,10]. Moreover, activated vitamin D treatment reduces all-cause and cardiovascular mortality rates in patients with CKD and those undergoing hemodialysis [11,12]. The critical role of the vitamin D endocrine system in disease prevention extends beyond the classic regulation of calcium and phosphorous homeostasis and skeletal integrity, to its potentially pleiotropic effects on extra-mineral metabolism, including kidney function.
This review focuses on the potential role of active vitamin D in the treatment of kidney disease beyond the correction of secondary hyperparathyroidism in patients with CKD. Therefore, we discuss altered vitamin D metabolism in kidney disease, and the potent renal protective mechanism of active vitamin D and its analogues in various animal models and patients with CKD.
ALTERED VITAMIN D METABOLISM IN KIDNEY DISEASE
Vitamin D is a steroid prohormone synthesized in the skin from cholesterol-derived precursors (i.e., 7-dehydrocholesterol) through a photochemical process. Alternatively, vitamin D can be obtained from various dietary sources such as fortified dairy products, fish oils, and mushrooms. Two native, biologically inactive, forms of vitamin D exist: vitamin D3 (cholecalciferol), which is derived from the skin and animal products, and vitamin D2 (ergocalciferol), which is of nonanimal origin [13]. These inactive forms are converted into their biologically active forms in the liver and kidney via two hydroxylation steps. The first step in the metabolic activation of vitamin D is metabolism to form 25-hydroxyvitamin D [25(OH)D] in the liver. This is the main storage and circulating form of vitamin D, and is therefore used as an indicator of vitamin D levels [14]. The second step occurs in the kidney, where 25(OH)D is converted to 1,25 dihydroxyvitamin D [1,25(OH)2D] by the enzyme 1α-hydroxylase, which is expressed primarily in the proximal tubular epithelial cells of the kidney [15]. However, this enzyme has been detected in extrarenal cells and tissues, including the prostate, breasts, colon, lungs, pancreatic β-cells, monocytes, endothelial cells, vascular smooth muscle cells, osteoblasts, and parathyroid cells [16]. This extra-renally produced 1,25(OH)2D primarily functions as an autocrine or paracrine factor at these sites, and may play a role in various nonclassical actions of vitamin D [15,17]. The final step in the vitamin D metabolic pathway is its inactivation, a process catalyzed by 24-hydroxylase, resulting in catabolism of 1,25(OH)2D and 25(OH)D into 1,24,25 trihydroxyvitamin D [1,24,25(OH)3D], and ultimately into water-soluble calcitroic acid and the inactive blood metabolite 24,25(OH)2D [13,18].
The kidney plays a central role in vitamin D metabolism and regulation of its circulating levels. Therefore, impaired renal function may lead to vitamin D deficiency, as has been observed in patients with CKD. There seem to be several mechanisms involved in decreased production of 1,25(OH)2D that occur over the course of CKD progression. A decrease in renal mass limits the amount of 1α-hydroxylase available for the production of the active vitamin D metabolite [19]. During the course of CKD, progressive decreases in GFR may also limit the delivery of 25(OH)D to 1α-hydroxylase, resulting in reduced production of 1,25(OH)2D [20]. However, even mild reductions in GFR cause 1,25(OH)2D levels to fall, suggesting that another mechanism may explain the decreased levels of vitamin D metabolites associated with kidney disease.
The 25(OH)D bound to its carrier, the vitamin D-binding protein, is filtered through the glomerulus and absorbed into the proximal tubules by the receptor megalin [21,22]. Megalin is an endocytic receptor also responsible for the renal tubular reabsorption of albumin and other low molecular weight proteins [22]. Interestingly, megalin knockout mice develop low molecular weight proteinuria and lose large amounts of vitamin D/vitamin D binding protein in their urine, resulting in severe vitamin D deficiency and bone disease, despite sufficient delivery of substrates to the kidney proximal tubules [22,23]. Renal megalin is induced by active vitamin D and the vitamin D receptor (VDR) complex [24]. Therefore, vitamin D deficiency could result from decreased megalin, creating a vicious cycle for 25(OH)D uptake and active vitamin D production.
Another key factor that can induce low vitamin D status over the course of kidney disease is the increase in fibroblast growth factor 23 (FGF-23) levels, which can directly suppress the activity and expression of 1α-hydroxylase [25,26]. Serum levels of FGF-23 increase with the decline in GFR and the increased phosphate level [27,28]. In addition, dietary phosphate intake decreases renal 1α-hydroxylase activity and expression of its messenger ribonucleic acid (mRNA), independently of changes in parathyroid hormone (PTH) and calcium levels [15,29]. Thus, persistent phosphate retention and increased FGF-23 due to declining kidney function may be a major mechanism contributing to the suppressed activation of 1α-hydroxylase, which decreases the levels of 1,25(OH)2D. FGF-23 also upregulates expression of 24-hydroxylase, which decreases the concentration of circulating 1,25(OH)2D [30]. In addition to these factors, the activity of 1α-hydroxylase may also be suppressed by N-terminally truncated PTH fragments [31] or progressive retention of uremic toxins [31,32]. An overview of how vitamin D metabolism is altered during the course of kidney disease is depicted in Fig. 1.
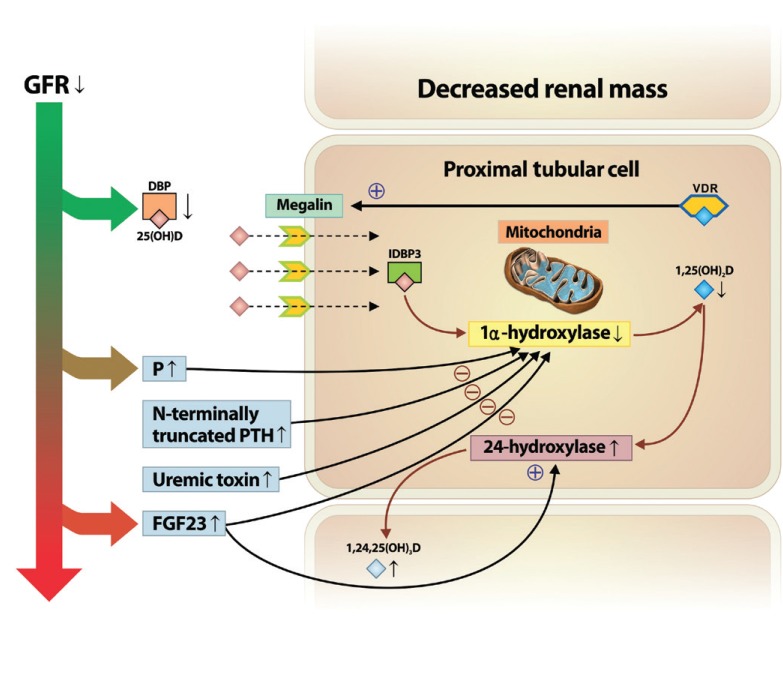
Altered vitamin D metabolism in the course of kidney disease progression. In chronic kidney disease progression, decreased renal mass limits the amount of 1α-hydroxylase in renal proximal tubular cells. In addition, decreases in glomerular filtration rate (GFR) and low megalin content contribute to impaired 25(OH)D uptake and protein reabsorption. Moreover, increased levels of serum level of phosphate (P), fibroblast growth factor 23 (FGF23), N-terminally truncated parathyroid hormone (PTH) fragments, and uremic toxins, along with kidney function decline may contribute to the suppressed activation of 1α-hydroxylase, resulting in decreased levels of 1,25(OH)2D. Also, increased FGF23 upregulates the expression of 24-hydroxylase, resulting in the catabolism of 1,25(OH)2D to the inactive form of vitamin D, 1,24,25(OH)3D. DBP, vitamin D binding protein; VDR, vitamin D receptor, IDBP3, intracellular vitamin D binding protein 3.
RENOPROTECTIVE MECHANISMS OF VITAMIN D IN EXPERIMENTAL STUDIES
Results from animal model studies have suggested the potential renoprotective effects of active vitamin D and its analogues (Table 1) [33,34,35,36,37,38,39,40,41,42,43,44,45]. Beyond its role in calcium and phosphate homeostasis, vitamin D is an important modulator of cellular proliferation, inflammation, differentiation, and immunity [33,46,47]. Vitamin D attenuates kidney injury by suppressing fibrosis, inflammation, and apoptosis, by inhibiting multiple pathways known to play a role in kidney injury, including the renin-angiotensin-aldosterone system (RAAS) [48,49,50], the nuclear factor-κB (NF-κB) [35,36,37,51], the transforming growth factor-β (TGF-β)/Smad [34,38,39,51,52], and the Wnt/β-catenin signaling pathways [40,51]. Moreover, paricalcitol (19-nor-1,25-dihydroxyvitamin D2) is an active, nonhypercalcemic vitamin D analogue that shows similar biological activity, with fewer adverse effects and increased tolerance, as compared to those of vitamin D [53].
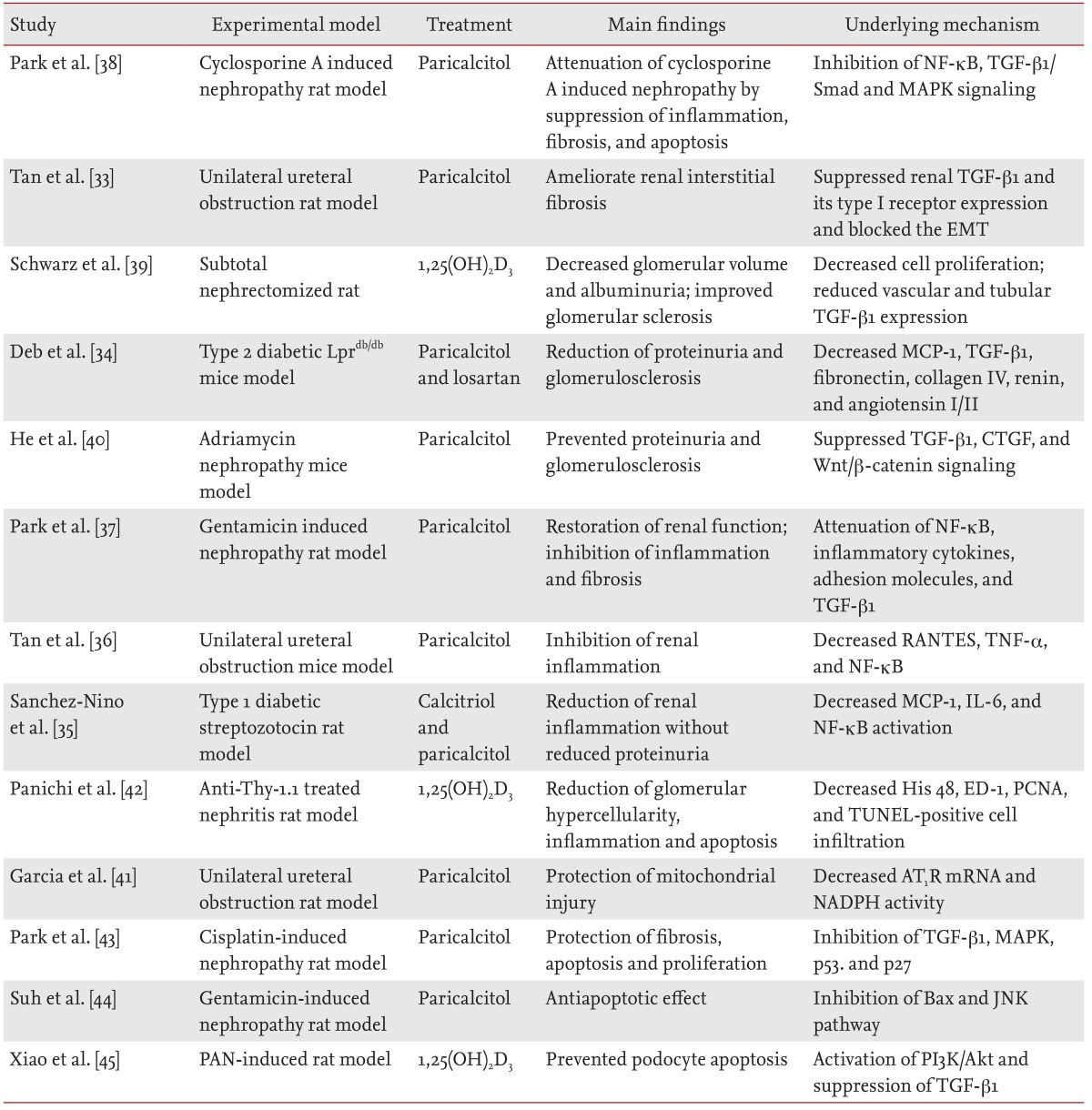
Summary of studies investigating the effects of vitamin D and vitamin D analogues in experimental models of kidney disease
Antifibrotic effects of vitamin D and its analogues
Progressive renal fibrosis is the final manifestation of CKD leading to irreversible loss of nephrons and ESRD [54]. Fibroblast activation and proliferation are triggered by locally secreted fibrogenic cytokines such as TGF-β1, connective tissue growth factor, and monocyte chemotactic protein-1 (MCP-1) [55,56]. Among these fibrogenic cytokines, TGF-β signaling initiates renal tubular cell proapoptotic effects and the epithelial-to-mesenchymal transition (EMT) in tubular epithelial cells. The EMT is a process in which renal tubular cells lose their epithelial phenotype and acquire new features of the mesenchyme [57]. We demonstrated that TGF-β1 expression increases in cyclosporine A (CsA)-treated rats, which was characterized by inflammatory cell infiltration, tubule-interstitial fibrosis, and glomerular sclerosis [38,58]. Paricalcitol treatment effectively prevented the TGF-β1-induced EMT and extracellular matrix accumulation, as evidenced by attenuated collagen deposition and fibrosis in CsA-treated rats. This result suggests that active vitamin D has antifibrotic effects [38]. We have also demonstrated that paricalcitol reduces expression of extracellular signal-regulated kinase 1/2 and c-Jun N-terminal kinase via TGF-β1 activation in CsA-treated rat kidneys [38]. Schwarz et al. [39] showed reduced albuminuria and suppression of glomerular sclerosis in subtotal nephrectomy rat models following treatment with 1,25(OH)2D3. Moreover, TGF-β1 expression was attenuated in vascular and tubular cells in 1,25(OH)2D3-treated rats [39]. In a mouse model of tubulointerstitial fibrosis that was induced by unilateral ureteral obstruction, paricalcitol reduced renal interstitial fibrosis and directly suppressed TGF-β1-mediated tubular EMT [52]. In addition, in a model of type 2 diabetes, the combination of paricalcitol and losartan appeared to synergistically reduce the extracellular matrix protein in the glomerulus by suppressing TGF-β1 and the RAAS [34].
A recent study showed that Wnt/β-catenin signaling, which can be inhibited by paricalcitol treatment, plays a critical role in promoting proteinuria and renal fibrosis [40]. Interestingly, our in vitro data indicate that the β-catenin signaling pathway may mediate 4-hydroxy-2-hexenal (HHE)-induced renal tubular EMT, which can be effectively inhibited by paricalcitol-induced VDR/β-catenin complex formation in human proximal tubular epithelial cells [51]. Paricalcitol inhibits β-catenin-mediated gene transcription by inducing VDR, resulting in sequestration of β-catenin transcriptional activity in the nuclei. Overall, the literature has demonstrated that active vitamin D and its analogues can ameliorate renal tubulointerstitial fibrosis or glomerulosclerosis.
Anti-inflammatory effects of vitamin D and its analogues
Renal inflammation is an essential pathological process in the evolution of CKD [59]. Several studies have shown the role of active vitamin D in the modulation of renal inflammation [37,38,41]. The anti-inflammatory properties of active vitamin D and its analogues may be attributed to their ability to suppress the NF-κB pathway, a key transcription factor that is thought to mediate acute and chronic inflammation and fibrogenesis by regulating gene expression of cytokines, chemokines, and adhesion molecules (including interleukin-6, MCP-1, and tumor necrosis factor-α) [60]. In this context, we demonstrated that paricalcitol protects against kidney injury by blocking NF-κB activity and reducing renal inflammation in CsA and gentamicin-induced renal injury [37,38]. Paricalcitol prevents upregulation of inflammatory cytokines and adhesion molecules in these experimental models. Tan et al. [36] showed that paricalcitol attenuates renal infiltration of inflammatory cells and inhibits tubular Regulated on Activation, Normal T cell Expressed and Secreted (RANTES) expression, which is an important proinflammatory chemokine in the obstructed kidney. This anti-inflammatory effect of paricalcitol seems to be mediated by inducing the complex formation of VDR and NF-κB p65 components, thereby reducing its binding to promoter regions of the target genes in vitro [36]. In addition, paricalcitol exerts an NF-κB-dependent anti-inflammatory effect on HHE-induced renal injury by inactivating the mitogen-activated protein kinase pathway [51]. In animal models of diabetic nephropathy, treatment with calcitriol and paricalcitol reduces infiltration of inflammatory cells and NF-κB activation in the glomerulus [35]. Similarly, administration of calcitriol attenuates glomerular hypercellularity and inflammatory infiltration in the anti-Thy-1.1 glomerulonephritis model [42]. Moreover, vitamin D may exert its anti-inflammatory influence through pleiotropic effects. Paricalcitol attenuates renin and angiotensin II expression in VDR knockout mice [50], suggesting that paricalcitol inhibits renal inflammation by suppressing the RAAS, as angiotensin II is a known proinflammatory stimulus. Taken together, these findings indicate that vitamin D and its analogues might be useful for treating inflammatory kidney diseases.
Antiapoptotic effects of vitamin D and its analogues
The decline in renal function during CKD results from renal cell death that precipitates functional and structural changes in the kidney [61]. Several common renal insults cause apoptosis in the kidney, including ischemia, toxic injury, radiation, and ureteral obstruction. Garcia et al. [41] reported that paricalcitol reduces the number of terminal deoxynucleotidyl transferase dUTP nick end labeling positive apoptotic cells and restores the high levels of angiotensin II type 1 receptor (AT1R) mRNA and NADPH activity in mitochondrial fractions in an obstructed kidney. This observation indicates that the antiapoptotic and AT1R-dependent protective effects occur at the mitochondrial level. Cisplatin is one of the most frequently used chemotherapeutic agents against solid tumors. However, cisplatin may activate proapoptotic genes and repress antiapoptotic genes through transcriptional regulation [62]. We showed that paricalcitol attenuates the increased expression of phospho-p53 and p21, which initiate apoptotic processes in a cisplatin-induced rat model [43]. We also reported that an increased Bax/Bcl-2 ratio and the cleaved form of caspase-3, which are apoptotic markers, are reversed by paricalcitol treatment in gentamicin-induced kidney injury [44]. In addition, 1,25(OH)2D3 prevents puromycin aminonucleoside-induced apoptosis of glomerular podocytes by activating the phosphatidylinositol 3-kinase/Akt-signaling pathway [45]. Conversely, a recent study demonstrated that increased activation of CYP24A1 induced by vitamin D supplementation would accelerate kidney apoptosis through increased caspase-3 expression, particularly in a diabetic experimental mice model [63]. Although active vitamin D and its analogues appear to have an antiapoptotic effect in various experimental nephropathy models, more studies are required to reveal the disease-specific differences of vitamin D metabolism.
VITAMIN D DEFICIENCY AS A CONTRIBUTOR TO THE DEVELOPMENT OF CKD
High prevalence of vitamin D deficiency or insufficiency in patients with CKD has been well documented in recent epidemiological studies. Despite the lack of consensus on optimal vitamin D levels, the recent Kidney Disease Improving Global Outcomes guidelines suggest definitions of vitamin D deficiency as serum 25(OH)D levels < 10 to 15 ng/mL (25 to 37 nmol/L) and insufficiency as ≥ 10 but < 20 to 32 ng/mL (50 to 80 nmol/L) [64]. However, many studies define serum 25(OH)D levels < 20 ng/mL as deficient, 20 to 29.9 ng/mL as insufficient, and ≥ 30 ng/mL as sufficient [6,65,66]. In a large population-based study, a serum 1,25(OH)2D decline was observed during early stages of CKD, before significant elevation in PTH or hyperphosphatemia [67]. In addition, the prevalence of deficiency appears to increase with the progression of kidney disease [68]. Vitamin D deficiency is associated with the short life expectancy of patients with ESRD. Treatment with active vitamin D and its analogues improves survival in the hemodialysis population [12,69,70,71]. Interestingly, a retrospective study showed that serum 25(OH)D is an independent inverse predictor of kidney disease progression and death in patients with earlier stages of CKD [72]. These clinical data suggest that the association between vitamin D therapy and reduced mortality in patients with CKD is beyond the effects of vitamin D on PTH and mineral metabolism. In a recent meta-analysis of prospective observational studies, Pilz et al. [10] estimated a significant 14% decrease in the relative risk of mortality per 10 ng/mL increase in 25(OH)D levels, indicating that higher vitamin D levels are associated with significantly improved survival in patients with CKD. However, there is no definite evidence for improved survival in patients with CKD being treated for low 25(OH)D levels using natural or active vitamin D supplementation. Therefore, further randomized controlled trials are needed to determine whether correcting vitamin D deficiency affects the development of ESRD or mortality in patients with CKD.
Improvement of proteinuria and renal inflammation is considered an important determinant of progression of cardiovascular and renal diseases [73,74]. Several studies have shown a correlation between vitamin D deficiency and an increased degree of albuminuria [75,76]. A recent large randomized controlled trial, the Vitamin D receptor Activator for Albuminuria Lowering study, confirmed that adding 2 µg paricalcitol for RAAS blockade reduces albuminuria and blood pressure in patients with diabetic nephropathy [77]. Moreover, in a recent meta-analysis, active vitamin D therapy with either paricalcitol or calcitriol provided a significant reduction of proteinuria in patients with CKD in addition to current use of RAAS blockade [78]. That study showed that proteinuria decreased by 16% in patients treated with active vitamin D, and increased by 6% in patients receiving the control treatment.
Vitamin D deficiency has been associated with a higher risk of cardiovascular disease in patients with CKD and the general population [79]. Treatment with active vitamin D attenuates myocardial hypertrophy in experimental models of cardiac hypertrophy and prevents the development of heart failure [80,81]. The Paricalcitol Capsule Benefits in Renal Failure Induced Cardiac Morbidity study was performed to evaluate the beneficial effects of paricalcitol on left ventricular hypertrophy in patients with stage 3 and 4 CKD [82]. Changes in the left ventricular mass index (LVMI), the primary endpoint, were not significantly different between the paricalcitol-treated and control groups, but cardiovascular-related hospitalization was lower in the paricalcitol-treated group. Similarly, a recent prospective randomized controlled trial showed that 52 weeks of treatment with oral paricalcitol did not regress LVMI or improve cardiac function in patients with stages 3 to 5 CKD [83]. One potential explanation for these results is that paricalcitol may have increased FGF-23 expression, which directly induces cardiomyopathy in animal models [84].
In contrast, evidence is accumulating that noncardiovascular mortality (including infection and malignancies) also increases in patients with CKD. In the NHANES III cohort study of 3,011 patients with CKD who were not on dialysis, vitamin D levels < 15 ng/mL were associated with a significant increase in noncardiovascular mortality, as compared to those with vitamin D levels < 30 ng/mL [9]. In addition, nutritional vitamin D has anti-inflammatory and immune-regulating properties. A recent small randomized controlled trial study showed that oral cholecalciferol supplementation decreases serum MCP-1 in patients with early CKD [85]. Table 2 summarizes the studies that have examined the effects of vitamin D and its analogues on proteinuria, mortality, and cardiovascular disease in patients with CKD and those undergoing dialysis [69,70,71,77,82,83,85,86].
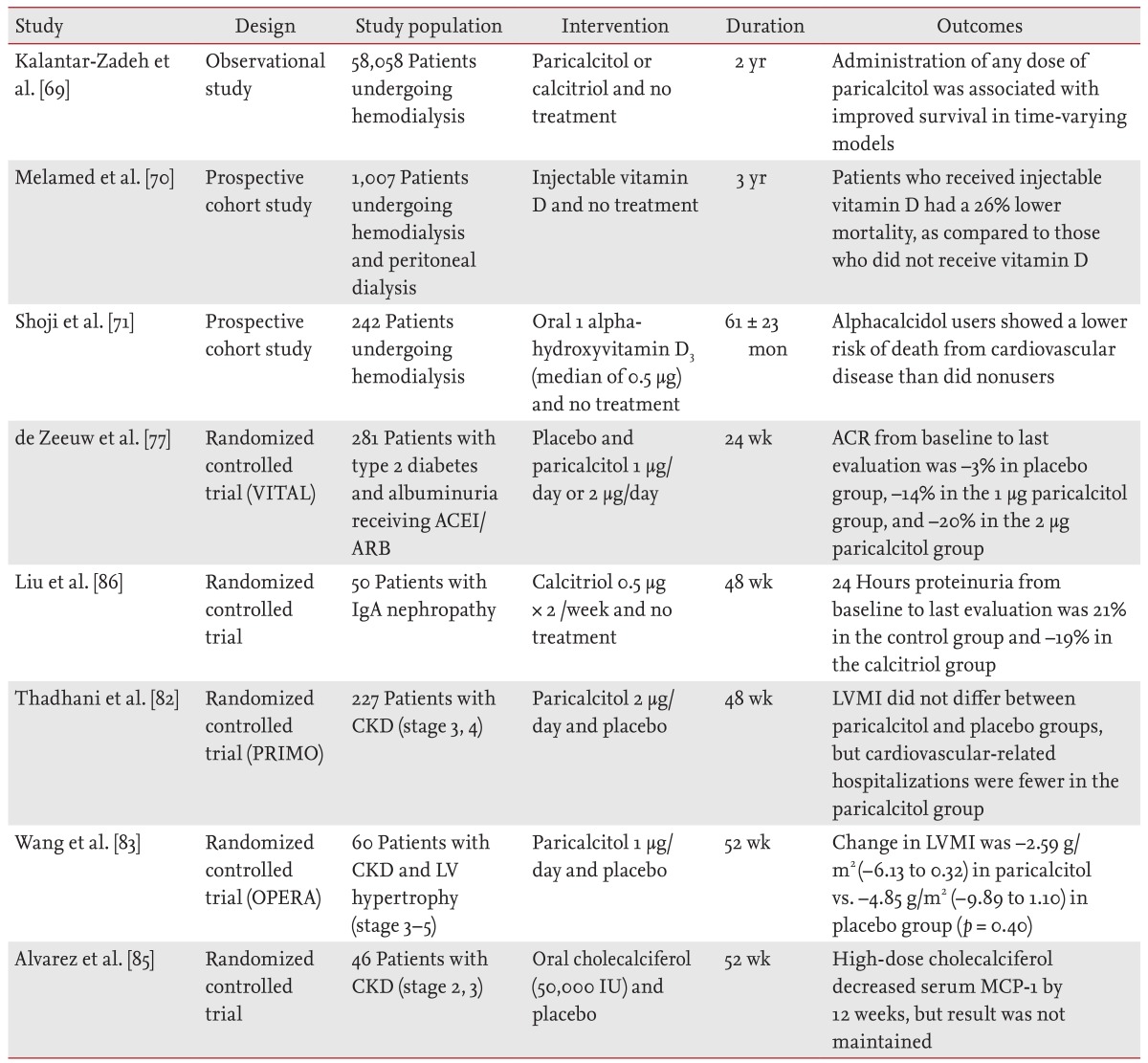
Summary of studies evaluating the effects of vitamin D and vitamin D analogues in patients with chronic kidney disease and dialysis
CONCLUSIONS
Progressive loss of kidney function is associated with considerable morbidity and mortality and is an important global health problem, given the high prevalence of CKD. The limitations of classic pharmacological therapy (i.e., RAAS blockade and statins) in delaying the progression of kidney injury have led to an increased interest in finding novel therapeutic agents that can stop the progression of kidney function loss or even improve kidney function. Treatment with active vitamin D or its analogues shows renoprotective effects by preventing fibrosis, apoptosis, and inflammation in various experimental models. Furthermore, vitamin D deficiency or insufficiency is a common condition in predialysis or dialysis patients with CKD, and serum levels of vitamin D appear to be inversely correlated with kidney function. Recent clinical studies report that the reduced proteinuria and mortality in CKD patients treated with active vitamin D is beyond the classical role of vitamin D in the maintenance of bone and mineral metabolism. Consequentially, vitamin D analogues, including paricalcitol, are emerging as potential treatment options for achieving better clinical outcomes in patients with advanced kidney disease and those who are undergoing hemodialysis.
However, current guidelines only recommend vitamin D treatment in patients with moderate CKD accompanied by secondary hyperparathyroidism and vitamin D insufficiency. Many issues must still be resolved, including testing the different vitamin D analogues, assessing whether nutritional vitamin D truly increases survival, and determining whether clinical outcomes vary according to disease-specific vitamin D metabolism. Therefore, further investigations are required to answer these questions and, thereby, uncover the potential renoprotective mechanisms of vitamin D treatment for patients with CKD.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT, and Future Planning (2013R1A2A2A01067611) and by the National Research Foundation of Korea grant (MRC for Gene Regulation, 2011-0030132) funded by the Korean government (MSIP).
Notes
No potential conflict of interest relevant to this article was reported.