Role of dipeptidyl peptidase-4 inhibitors in new-onset diabetes after transplantation
Article information
Abstract
Despite strict pre- and post-transplantation screening, the incidence of new-onset diabetes after transplantation (NODAT) remains as high as 60%. This complication affects the risk of cardiovascular events and patient and graft survival rates. Thus, reducing the impact of NODAT could improve overall transplant success. The pathogenesis of NODAT is multifactorial, and both modifiable and nonmodifiable risk factors have been implicated. Monitoring and controlling the blood glucose profile, implementing multidisciplinary care, performing lifestyle modifications, using a modified immunosuppressive regimen, administering anti-metabolite agents, and taking a conventional antidiabetic approach may diminish the incidence of NODAT. In addition to these preventive strategies, inhibition of dipeptidyl peptidase-4 (DPP4) by the gliptin family of drugs has recently gained considerable interest as therapy for type 2 diabetes mellitus and NODAT. This review focuses on the role of DPP4 inhibitors and discusses recent literature regarding management of NODAT.
INTRODUCTION
New-onset diabetes after transplantation (NODAT) is a serious and common complication after solid organ transplantation. This clinical dilemma increases the risk of cardiovascular disease, infection (cytomegalovirus and hepatitis C virus), and graft damage (graft rejection and loss) and decreases the patient and graft survival rates. Additionally, rejection of the graft affects the incidence of NODAT, resulting in a vicious circle [1-4]. Despite strict pre- and post-transplantation screening, the incidence of NODAT remains extremely high. NODAT reportedly occurs in 2.5% to 44.2% of liver transplant recipients [5,6], 4% to 40% of heart transplant recipients [7], and 30% to 47% of lung transplant recipients [8,9]. A multicenter observational study of 527 kidney transplant recipients (KTRs) reported that the incidence NODAT is 5.5% and 8.4% at 1 and 2 years post-transplantation, respectively [10]. Another long-term study showed that 60.2% of KTRs developed maintenance NODAT and 54.7% of KTRs manifested transient post-transplantation hyperglycemia among 176 KTRs from 2001 to 2012 [11]. The overall incidence of NODAT in patients who undergo solid organ transplantation is 2% to 60%. This wide variation is dependent on each study’s definition of NODAT, which is based on different diagnostic criteria, observation periods, presence of risk factors, and types of immunosuppressants used.
Multiple risk factors are associated with the development of NODAT and are broadly classified into two categories: (1) nonmodifiable risk factors, including old age (> 40 years) [12,13], ethnicity (African-American and Hispanic) [14], positive family history of diabetes mellitus (DM) [13], human leukocyte antigen mismatch, donor source, occurrence of an acute rejection episode, genetic factors, and autosomal dominant polycystic kidney disease [15,16]; (2) modifiable risk factors, including individualized immunosuppressants (tacrolimus, corticosteroids, and sirolimus) [17,18], obesity (body mass index ≥ 30 kg/m2) or other components of metabolic syndrome [19], viral infections (cytomegalovirus and hepatitis C virus) [20-22], and peritoneal dialysis [23]. The NODAT definition appears to be important for delineating preventive strategies. In 2003, the World Health Organization and the American Diabetes Association refined the NODAT definition based on three criteria [24]: in addition to symptoms of DM, the patient must have a casual plasma glucose concentration of ≥ 200 mg/dL (11.1 mmol/L), fasting plasma glucose concentration of ≥ 126 mg/dL (7.0 mmol/L), or a 2-hour plasma glucose (2HPG) concentration of ≥ 200 mg/dL (11.1 mmol/L) during an oral glucose tolerance test. The 2009 Kidney Disease Improving Global Outcomes clinical practice guidelines added hemoglobin A1c (HbA1c) as a screening criterion for diagnosing NODAT [25].
Management of NODAT requires a multifaceted approach because it affects multiple organs and the allograft itself. Pre- and post-transplantation screening based on the glucose profile or glycosylated hemoglobin is strongly recommended. Strict control to maintain normoglycemia remains the mainstay of treatment for transplant recipients who develop DM with overt micro- and macroalbuminuria. In addition, use of renin-angiotensin system blockers and switching immunosuppressants to more adequate regimens are effective for minimizing the impact of NODAT [26].
Dipeptidyl peptidase-4 (DPP4) inhibitors are selective inhibitors of DPP4, which is the key enzyme that regulates degradation of the two major incretins glucagon-like peptide-1 (GLP-1) and glucose inhibitory peptide (GIP). Therefore, DPP-4 inhibitors are now widely used to treat type 2 diabetes mellitus (T2DM) without weight gain or hypoglycemic risk. However, DPP4 is a serine protease that cleaves incretins and many other non-incretin peptides. Therefore, although they may be used for glucose control, DPP4 inhibitors may have pleiotropic effects, such as anti-inflammatory, antiapoptotic, and immunomodulatory actions. The protective effects of DPP4 inhibitors are mirrored in various renal injuries [27-29], DM [30,31], hepatic impairment [32], and cardiovascular disease models [33,34]. Using a well-known animal model, we recently demonstrated that the DPP4 inhibitor MK0626 protects against tacrolimus-induced pancreatic islet and renal injury via antiapoptotic and antioxidative actions [35,36]. In this review, we searched the literature for the pleiotropic roles of DPP4 in the prevention and management of NODAT and its comorbidities.
MOLECULAR BIOLOGY OF DPP4
DPP4 was first discovered by Hopsu-Havu and Glenner [37] in 1966. This protein is also called CD26 and is a ubiquitously expressed 110-kDa glycoprotein that belongs to the type 2 transmembrane protein family [38]. As a member of the serine peptidase/prolyl oligopeptidase family, DPP4 is often subclassified based on its structure and function as follows: membrane-bound peptidase (fibroblast activation protein (FAP)/seprase), resident cytoplasmic enzyme (DPP8 and DPP9), and nonenzymatic member (DPP6 and DPP10). These proteins share a typical α/β-hydrolase fold. DPP4 comprises four domains: a short cytoplasmic domain, a transmembrane domain, a flexible stalk segment, and the extracellular domain, which is further separated by a glycosylated region, the cysteine-rich region, and the catalytic region [38,39]. DPP4 can cleave dozens of peptides, including chemokines, neuropeptides, and regulatory peptides, containing a proline or alanine residue at position 2 of the amino-terminal region [40]. Despite the preference for proline at position 2, alternate residues at the penultimate position are also cleaved by DPP4, indicating a required stereochemistry for cleavage. This DPP4 cleavage at post-proline peptide bonds inactivates peptides and/or generates new bioactive peptides, thereby regulating diverse biological processes.
Most of the in vivo and in vitro experimental approaches used in this context have been employed to identify and characterize DPP4 substrates by incubation with plasma containing DPP4, transfected DPP4, or purified soluble DPP4. The results of these studies have shown that DPP4 substrates can be broadly classified into physiological and pharmacological substrates, the former of which include GIP and GLP-1 and the latter of which consist of a superfamily member, such as brain natriuretic peptide, erythropoietin, endomorphin-1, or glucagon [38-40]. Because of its diverse substrates, DPP4 exerts pleiotropic actions via protease activity, associations with adenosine deaminases, interactions with the extracellular matrix, cell surface co-receptor activity, and regulation of intracellular signal transduction coupled to the control of cell migration and proliferation. Thus, DPP4 triggers multiple biological activities in paracrine or endocrine manners.
PIVOTAL DPP4 SUBSTRATES
Numerous peptides that contain a cleavable amino acid sequence at their penultimate position are potential DPP4 substrates. There seems to be a size limitation, at least for cytokines, because DPP4 is more prone to cleave substrates with approximately 24 amino acids [38,39]. The incretin hormones are secreted from the gut and account for approximately 50% of the insulin secretion that occurs within minutes after a meal. These hormones stimulate insulin secretion and suppress glucagon release by binding to its distinct receptors on pancreatic β-cells. GIP and GLP-1 are the most potent glucose-lowering hormones, and both proteins belong to the same glucagon peptide superfamily and share amino acid characteristics [40]. GIP is a 42-amino acid peptide derived from preproGIP via post-translational processing by prohormone convertase (PC) 1/3, which originates mainly from enteroendocrine K cells [41,42]. GLP-1 is secreted from L cells of the distal gut after post-translational cleavage of proglucagon by PC 1/3 in the bloodstream; DPP4 can cleave GLP-1 [43]. Intact GLP-1 promotes glucose-stimulated insulin secretion and suppresses glucagon secretion, appetite, and gastric emptying via the GLP-1 receptor (GLP-1R) [41]. DPP4 cleavage eliminates the classical glucoregulatory actions of GLP-1 and generates peptides with a 100-fold lower receptor affinity, illustrating that the N-terminal residues are required for engaging GLP-1R. GIP is also expressed in islet α-cells and stimulates insulin secretion [44]. DPP4 cleaves GIP to release the dipeptide (TyrAla); however, GIP is unable to activate the GIP receptor and functions as an antagonist in vitro. Unlike GLP-1, GIP has no effect on glucagon secretion, but regulates fat metabolism in adipocytes.
DPP4 INHIBITORS
The majority of DPP4 substrates are so-called incretin hormones, which are key regulators of postprandial insulin release. Inhibiting DPP4 may result in its greater bioavailability, thereby prolonging the half-life of insulin action. Thus, DPP4 inhibitors have been approved for treating T2DM, either as a monotherapy, add-on, or combined therapy with other glucose-lowering agents. In addition to the lack of an effect on satiety and gastric emptying, the benefits of DPP4 inhibitors are their indifference to body weight gain and the risk of hypoglycemia. Five gliptins have been approved for clinical use: sitagliptin, vildagliptin, saxagliptin, linagliptin, and alogliptin. In addition, teneligliptin, anagliptin, and trelagliptin have been approved in Japan and Korea. This article outlines the five gliptins that are commonly used in clinical practice.
Sitagliptin was the first DPP4 inhibitor approved in 2006 for clinical use to treat T2DM and is currently available as a monotherapy or fixed-dose combination with other antidiabetic agents, such as metformin [45]. It is a competitive and fully reversible DPP4 inhibitor that has a half-maximal inhibitory concentration (IC50) of 18 nM and interacts with the S2 extensive subsite of the DPP4 active center [46]. Its high selectivity ensures targeted action on DPP4 and avoids unwanted secondary effects or potential toxicities resulting from cross-inhibition of other DPP peptides, such as DPP8 or DPP9 [47]. Sitagliptin (50 mg once daily) may reduce DPP4 activity by 80% within 12 hours, and 100 mg of sitagliptin maintains similar effectiveness for 24 hours [45]. Moreover, sitagliptin has high bioavailability, and approximately 80% of the parent drug is excreted unchanged in the urine. Therefore, no dose adjustment is needed in patients with mild renal insufficiency (creatinine clearance > 50 mL/min). However, a half dose (50 mg) or a one-quarter dose (25 mg) is recommended for patients with moderate (creatinine clearance of 30 to 50 mL/min) or severe (creatinine clearance of < 30 mL/min) renal insufficiency [48].
In contrast to sitagliptin, vildagliptin only binds to the S1 and S2 subsites and forms a covalent bond with the nitrile group of their cyanopyrrolidine moiety and Ser630 of DPP4. Because it is a substrate-enzyme blocker, vildagliptin has lower DPP4 selectivity (IC50 = 100 nM) than does sitagliptin and cross-inhibits DPP8 [49]. Orally administered vildagliptin is well tolerated, rapidly absorbed (within 3 hours), and mainly metabolized by the liver and partially by the kidney (27%) [50]. Although the major route for vildagliptin excretion is the liver, no difference in excretion is observed in patients with mild, moderate, or severe hepatic impairment, suggesting that no dose adjustment is necessary for hepatically impaired patients [50]. In contrast, the recommended dose vildagliptin is the half dose (50 mg daily) for patients with moderate or severe renal insufficiency or end-stage renal disease but not in patients with mild renal impairment.
Similar to vildagliptin, saxagliptin is a selective and reversible DPP4 inhibitor that binds to the S1 and S2 subsites. However, it differs from other gliptins because it has an active metabolite (5-hydroxy-saxagliptin, BMS-510849) that is also a selective, reversible, and competitive DPP4 inhibitor. Both the parent form (12% to 29%) and the saxagliptin metabolite (21% to 52%) can be secreted by the kidneys [51]. As a result, the saxagliptin dose should be reduced by 50% (2.5 mg daily) in patients with moderate or severe renal impairment.
Linagliptin was approved in 2011 by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency for treating T2DM. This drug interacts with both the S1’ and S2’ subsites; thus, it has an 8-fold higher activity than other gliptins. Linagliptin binds tightly to plasma proteins after oral administration, and its pharmacokinetics are influenced by storable high-affinity binding to DPP4 in the plasma and tissues, leading to a long terminal half-life [52,53]. Linagliptin kinetics may be unaffected by food intake, as it is mainly excreted unchanged in the feces (> 84%). Recent clinical trials have shown that a multiple dose of linagliptin [54] or linagliptin combined with metformin [55] is safe and well tolerated compared with placebo, suggesting that linagliptin should be administered to patients with T2DM as either monotherapy or in combination with other antihyperglycemic agents without adjusting the dose.
Alogliptin was first approved by the Pharmaceuticals and Medical Devices Agency of Japan in 2010 and by the FDA in 2013 for treating T2DM. It is a potent and highly selective inhibitor of DPP4 with a mean IC50 of 6.9 nM and 1,000-fold increased selectivity for DPP4 compared with that of the closely related serine proteases DPP2, DPP8, DPP9, FAP/seprase, prolyl endopeptidase, and tryptase [56]. Alogliptin exhibits favorable pharmacokinetic, pharmacodynamic, and pharmacologic safety profiles. Therefore, alogliptin as a monotherapy or addon to metformin, pioglitazone, glipizide, glibenclamide, voglibose, or insulin significantly improves glycemic control compared with placebo or active comparators in adult and elderly patients with inadequately controlled T2DM [57,58]. Because the kidney is the main excretion route for alogliptin, accounting for 60% to 71% [58] of excretion, the oral dose should be reduced or withdrawn in patients with renal impairment. The details of DPP4 inhibitors are summarized in Table 1 [45,50,56,58-64].
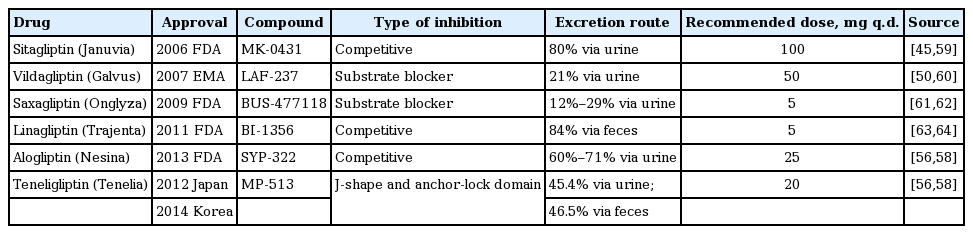
Outline of common dipeptidyl peptidase-4 inhibitors
ANTIDIABETIC EFFECT OF DPP4 INHIBITORS
Transplant-associated hyperglycemia comprises NODAT, impaired fasting glucose, and impaired glucose tolerance, all of which are closely related to increased morbidity and mortality in KTRs. Although NODAT confers a high risk for premature allograft failure and increased cardiovascular events, therapeutic strategies for this condition remain underexplored. Metformin is the first-line agent of choice for treating T2DM in the general population. However, the use of metformin in KTRs is often limited because of concern about lactic acidosis. DPP4 inhibitors are a class of oral antidiabetic drugs that stabilize GLP-1 and GIP, resulting in improved glycemic control, reduced postprandial hyperglycemia, and a lower risk of weight-neutral and -lowering hypoglycemia in patients with T2DM. Overwhelming evidence shows that DPP4 inhibitors are effective for managing NODAT. Strom Halden et al. [65] reported that 50 to 100 mg/day of sitagliptin increased the median first- and second-phase insulin secretion rates by 56.3% and 39.3%, respectively, and significantly reduced fasting and 2HPG concentrations by 14.8 and 47.5 mg/dL, respectively, compared with those in a sitagliptin-free group of stable renal recipients with NODAT. Haidinger et al. [66] demonstrated that vildagliptin profoundly reduced the concentrations of HbA1c (6.1% vs. 6.5%) and 2HPG (182.7 mg/dL vs. 231.2 mg/dL) compared with placebo, which was almost achieved at the primary endpoint. Treatment with sitagliptin or vildagliptin had good efficacy and safety in both study arms, and associated adverse events were mild and appeared to be negligible. This concept is supported by studies [67-69] reporting similar efficacy and safety of DPP4 inhibitors for treating NODAT. DPP4 inhibitors are considered a novel treatment alternative for KTRs with overt NODAT.
ANTIHYPERTENSIVE EFFECT OF DPP4 INHIBITORS
Hypertension is an important cause of chronic kidney disease and a common complication of KTRs, accounting for 50% to 90% of their incidence [25]. Ogawa et al. [70] reported that an alternate-day treatment with sitagliptin significantly lowered systolic blood pressure (from 130.0 to 119.7 mmHg) and HbA1c levels in Japanese hypertensive patients with T2DM. However, their body mass index remained unchanged, and no association was found between systolic blood pressure and HbA1c level. The hypotensive effect of sitagliptin has also been observed in nondiabetic patients with mild to moderate hypertension, in whom both systolic and diastolic blood pressures decreased markedly after 5 days of sitagliptin treatment [71]. These clinical observations were further confirmed by animal studies using Zucker Diabetic Fatty rats [72] and spontaneously hypertensive rats [73], in which the antihypertensive effect of the DPP4 inhibitors in which urinary flow and sodium excretion increased due to decreased expression of the type 3 sodium-hydrogen transporter in the renal proximal tubule. The molecular mechanism underlying the antihypertensive effect of DPP4 inhibitors is multifactorial and may involve neuropeptide Y (NPY) and peptide YY (PYY). Because NPY and PYY are agonists of the endogenous Y (1) receptor, which mediates vasoconstriction, these peptides are cleaved by DPP4 to NPY (3–36) and PYY (3–36) [74,75]. This additional antihypertensive effect can extend the clinical use of DPP4 inhibitors to KTRs and patients with NODAT.
ANTI-INFLAMMATORY EFFECT OF DPP4 INHIBITORS
DM is a low-grade systemic inflammatory disease. Suppressing inflammation slows the progression of DM. In addition to preserving glucose homeostasis, DPP4 inhibitors exert pleiotropic actions, such as anti-inflammatory effects. Alogliptin inhibits Toll-like receptor-4-mediated extracellular matrix signal-regulated kinase (ERK) activation and ERK-dependent matrix metalloproteinase expression in U937 histiocytes [76]. Des-fluoro-sitagliptin (sitagliptin analog) markedly enhances GLP-1-induced cytosolic levels of cyclic adenosine monophosphate (cAMP) compared with GLP-1 alone in cultured human macrophages and endothelial cells, resulting in inhibition of nuclear factor-κB p65 nuclear translocation via the cAMP/protein kinase A pathway; it also suppresses production of the proinflammatory cytokines interleukin-1β (IL-1β), IL-6, tumor necrosis factor-α, and monocyte chemoattractant protein-1 in response to lipopolysaccharide (LPS) [77]. DPP4 inhibitors reduce cyclooxygenase-2, IL-1β, macrophage inflammatory protein-2, and TLR-4-mediated IL-6 expression in Zucker Diabetic Fatty rat [78], diabetic apolipoprotein E-deficient mice [34], and C57BL/6J-obese/obese mice [79], which parallels recovery from disease. Matsubara et al. [80] reported that sitagliptin significantly decreases high sensitivity C-reactive protein levels and improves endothelial function in human patients with uncontrolled DM. It is speculated that the anti-inflammatory properties of DPP4 inhibitors may be largely beneficial for KTRs with DM.
ANTIAPOPTOTIC EFFECT OF DPP4 INHIBITORS
Apoptosis is an active cell clearance mechanism that plays an important role in regulating cell numbers during homeostasis, development, and under disease conditions [81]. Although apoptosis is beneficial, it can also be deleterious if a critical number of resident cells are lost. In fact, the pathogenic role of apoptosis has been well described for a wide range of diseases, including DM and DM-associated micro- and macrovascular complications [82-84]. DM induces pancreatic β-cell apoptosis in vivo [85] and in vitro [86], and these cells are regulated by oxidative stress toward apoptotic cell death. Shimizu et al. [87] showed that vildagliptin increases pancreatic β-cell mass, improves aggravated endoplasmic reticulum stress, and restores pancreatic and duodenal homeobox 1 expression in diabetic pancreatic β-cell specific C/EBPB transgenic mice. The antiapoptotic effect of DPP4 inhibitors was also observed in studies of cardioprotection [88] and renoprotection [27] via modulation of the Bax to Bcl-2 ratio and caspase-3 activity. We recently reported that the DPP4 inhibitor MK-0626 attenuates both pancreatic and renal cell apoptosis in tacrolimus-induced diabetic rats and that this is associated with the regulation of 8-hydroxy-2’-deoxyguanosine, heme oxygenase-1, and manganese superoxide dismutase by preserving GLP-1 (Figs. 1 and 2) [35,36]. Our findings are consistent with those of a study performed by Chang et al. [89], which showed a role for sitagliptin in apoptosis and oxidative stress (glutathione peroxidase and malondialdehyde), favoring cell survival in a rat model of cardiac ischemia-reperfusion. Based on our findings and those of others, we speculate that DPP4 inhibitors trigger an antiapoptotic effect, partially by inhibiting oxidative stress injury.

Effect of MK-0626 on apoptosis and islet viability in tacrolimus-induced pancreatic and renal injured experimental rats. (Aa, Ba, Ca) In situ TdT-mediated dUTP-biotin nick end labeling (TUNEL) assay in pancreatic islets. (Ab, Bb, Cb) Acridine orange/propidium iodide staining of isolated islets. (Ac, Bc, Cc) TUNEL assay in renal tissues. The tacrolimus group (B) combined with the MK-0626 group (C) reduced apoptosis. (A) is the vehicle group (×400). Adapted from Lim et al. [36], with permission from Nature Publishing Group and Jin et al. [35].

Effect of MK-0626 on oxidative stress and apoptotic gene expression in tacrolimus (TAC)-induced pancreatic and renal injured experimental rats. (A) Immunoblot analysis of manganese superoxide dismutase (MnSOD), heme oxygenase-1 (HO-1), Bcl-2, active caspase-3, and β-actin. 8-Hydroxy-2'-deoxyguanosine (8-OHdG) levels in serum (B) and 24-hour urine (C). Adapted from Lim et al. [36], with permission from Nature Publishing Group and Jin et al. [35]. ap < 0.05 vs. vehicle (VH) group or VH + M groups; bp < 0.05 vs. TAC group.
IMMUNOMODULATORY EFFECT OF DPP4 INHIBITORS
Regardless of the above-mentioned effects, DM (particularly type 1 DM, autoimmune disease) is closely associated with immunological injury in which pancreatic β-cells are selectively destroyed by the immune system. Therefore, the inhibition provided by DPP4 may exert an immunomodulatory effect against DM because DPP4 is ubiquitously expressed in numerous cell types. In this context, whether DPP4 inhibitors possess immunomodulatory properties remains controversial. Sitagliptin (100 mg/day) administered to healthy volunteers [90] and patients with T2DM [91] for 28 days and 6 months showed that neither the systemic immune function (chemokine/cytokine release by stimulation with either LPS or anti-CD3) nor CD4+ T-cell activation are affected. Anz et al. [92] reported that sitagliptin, vildagliptin, and saxagliptin have no effect on the innate immune response in terms of cytokine secretion, immune cell activation, or lymphocyte trafficking after toll-like receptor ligand stimulation. In contrast, treatment of nonobese diabetic mice with MK0431 before and after islet transplantation reduces the effect of autoimmunity on graft survival by decreasing homing of CD4+ T-cells via cAMP/PKA/Rac1 activation [93]. Furthermore, linagliptin and DA-1229 reduce the onset of DM and the total mass of lymphocyte insulitis and protect the β-cell mass and neogenesis in nonobese diabetic and streptozotocin-induced mice [94,95]. The reasons for this discrepancy are unknown but may be related to the study setting and type of DM. Further studies are needed to resolve this issue.
CONCLUSIONS
DPP4 inhibitors were developed initially and approved for treating T2DM, based on inhibiting degradation of GLP-1 and GIP. Increasing evidence demonstrates that DPP4 inhibitors exert potential pleiotropic effects including anti-inflammation, antihypertension, antiapoptosis, and immunomodulation on the heart, vessels, and kidney, independent of their hypoglycemic effect (Fig. 3). Preclinical and clinical studies have shown that DPP4 inhibitors are well tolerated, safe, and efficacious and lower the risk of hypoglycemia in stable KTRs with NODAT. This is of great clinical relevance because of the huge proportion of transplant recipients with DM. The cardioprotective and renoprotective effects of DPP4 inhibitors offer an additional therapeutic avenue for this new drug class.
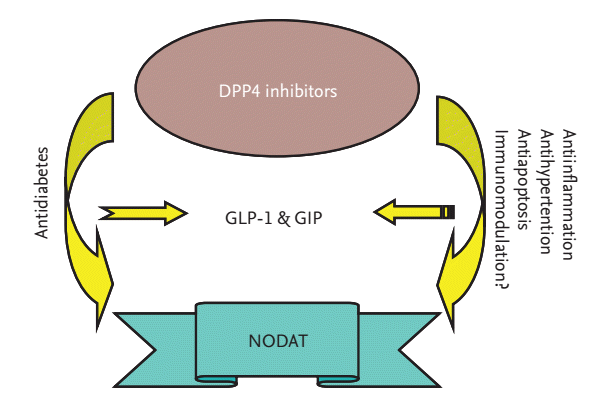
Paradigm of dipeptidyl peptidase-4 (DPP4) inhibitors actions on new-onset diabetes after transplantation (NODAT). DPP4 inhibitors exert antidiabetic effect dependent on the regulation of glucagon-like peptide-1 (GLP-1) and/or glucose inhibitory peptide (GIP) pathway. However, DPP4 inhibitors may also exert pleiotropic actions dependent or independent on GLP-1 and/or GIP pathway.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81160092).