Compound K attenuates glucose intolerance and hepatic steatosis through AMPK-dependent pathways in type 2 diabetic OLETF rats
Article information
Abstract
Background/Aims
Non-alcoholic fatty liver disease is associated with insulin resistance. Compound K (CK) is the final metabolite of panaxadiol ginsenosides that have been shown to exert antidiabetic effects. However, the molecular mechanism of the antidiabetic effects in the liver have not been elucidated; further, whether CK has beneficial effects in hepatosteatosis remains unclear. Therefore, we evaluated the effect of CK on hepatosteatosis as well as its mechanism in high-fat diet (HFD)-fed type 2 diabetic Otsuka Long-Evans Tokushima Fatty (OLETF) rats.
Methods
Twenty-four-week-old male OLETF rats were assigned to four groups: control (saline), CK 10 mg/kg, CK 25 mg/kg, or metformin 300 mg/kg (positive control); all treatments were administered orally for 12 weeks.
Results
Fasting glucose levels of the CK25 group were significantly lower than those of the control group during the 12 weeks. The results of the oral glucose tolerance test showed that both the glucose concentration after glucose loading and the fasting insulin levels of the CK25 group were significantly lower than those of the control. Hepatosteatosis was significantly improved by CK25. CK25 and metformin significantly increased the phosphorylation of hepatic adenosine monophosphate-activated protein kinase (AMPK). CK25 significantly inhibited the expression of sterol regulatory element-binding protein-1c and fatty acid synthase, while upregulating that of peroxisome proliferator-activated receptor-α and carnitine palmitoyltransferase-1.
Conclusions
CK improved glucose intolerance and hepatosteatosis in HFD-fed OLETF rats through AMPK activation, which has dual mode of action that involves decreasing the synthesis of fatty acids and increasing fatty acid oxidation.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is an important manifestation of metabolic syndrome, which includes insulin resistance, hypertension, central obesity, and dyslipidemia. Epidemiological studies and clinical trials have established the correlation between the increasing incidence of NAFLD and the increasing epidemics of obesity and type 2 diabetes mellitus [1]. NAFLD includes a wide spectrum of hepatic abnormalities such as fatty liver (steatosis), inflammation (hepatitis), cirrhosis, and carcinomas. Non-alcoholic steatohepatitis (NASH) is an intermediate stage that is characterized histologically by hepatic steatosis, lobular inflammation, and hepatocellular ballooning [2]. Although the exact mechanism of NASH is not clear, insulin resistance and oxidative stress have been implicated as key factors contributing to the progression of NASH. Several studies have demonstrated the beneficial effect of insulin sensitizers in NASH, including thiazolidinediones, biguanide, and antioxidants such as vitamin E and α-lipoic acid [3].
Adenosine monophosphate-activated protein kinase (AMPK) plays a key role in glucose metabolism including glucose transport, gluconeogenesis, and lipolysis. AMPK reduces fatty acid synthesis by decreasing the expression of sterol regulatory element-binding protein-1c (SREBP-1c) [4] and enhances fatty acid oxidation by increasing peroxisome proliferator-activated receptor-α (PPAR-α), PPAR-γ coactivator-1α, carnitine palmitoyltransferase-1 (CPT-1), and uncoupling proteins [5]. Therefore, AMPK can be a therapeutic target for insulin resistance.
Compound K (CK) is the final metabolite of panaxadiol ginsenosides [6]. CK has antitumor, apoptotic, and antidiabetic effects; the latter is responsible for stimulating insulin secretion in pancreatic β-cells and enhancing insulin sensitivity in hepatocytes and myocytes [7]. However, the effect of CK on NAFLD in conditions such as obesity and diabetic is unknown. In this study, the effect of CK on hepatosteatosis and its mechanism were investigated in high-fat diet (HFD)-fed type 2 diabetic Otsuka Long-Evans Tokushima Fatty (OLETF) rats.
METHODS
Animals
Twenty-four-week-old, male type 2 diabetic model OLETF rats were kindly provided by Otsuka Pharmaceuticals (Tokushima, Japan). The rats were individually housed and allowed free access to food and tap water under strictly controlled and pathogen-free conditions (room temperature, 23°C ± 1°C; relative humidity, 50% ± 10%; light cycle, 7:00 AM to 7:00 PM). The rats were fed a standard rodent pellet chow and acclimatized to their environment for 2 weeks prior to the commencement of the experiments. Thirty-two rats were randomly assigned to four subgroups and then treated daily with CK, metformin, or vehicle for 12 weeks: (1) control (n = 6) received vehicle (saline); (2) CK 10 mg/kg (CK10; n = 10); (3) CK 25 mg/kg (CK25; n = 10); and (4) metformin 300 mg/kg (n = 6) as a positive control. CK in the form of a dried powder was dissolved in saline and was orally administered by gavage daily. All treatments were orally administered. To induce hepatic steatosis, all rats were fed 60% HFD (Joongang Animal Laboratory, Seoul, Korea). The experimental procedures were conducted according to the animal care guidelines of the United States National Institutes of Health (NIH) and approved by the Animal Care and Use Committee of Kyung Hee University (IACUC approval No.2012-0002). The use of animals in the study was approved by the institution’s Animal Ethics Committee in accordance with Article 14 of the Korean Animal Protection Law or its equivalent. The use of animals in the study complied with Article 13 of the Korean Animal Protection Law (the principles for animal use) and relevant institutional policies.
Functional protocol
The body weight of each rat was measured at the beginning of the experiment and every week thereafter. Food intake was determined by measuring the difference between the pre-weighed chows and the weight of the food that remained every 24 hours during the first and last week of the 3-month diet period. Blood samples were collected from the tail vein every week of treatment and the serum was stored at –20°C until analysis. The fasting blood glucose level was assessed using an Accu-Chek active glucometer (Roche, Basel, Switzerland). After the 12- week treatment, whole blood was collected by inferior vena cava puncture from all rats. Each blood sample was held at room temperature for 1 hour. Serum was then collected by centrifugation at 4,000 rpm for 10 minutes and stored at –70°C until analysis. Glycated hemoglobin (HbA1c) levels were measured using a DCA Vantage Analyzer (Siemens, Tarrytown, NY, USA). Serum insulin was assayed by enzyme-linked immunosorbent assay (Millipore, Billerica, MA, USA). The oral glucose tolerance test (OGTT) was performed with rats that were fasted for 16 hours at 0 weeks and at 12 weeks. Glucose (2 g/kg body weight) was orally administered to the rats and blood samples were collected 0, 30, 60, 90, and 120 minutes after glucose loading.
Histological and biological analysis
The livers were obtained from each rat after perfusion of phosphate buffered saline (PBS). They were then fixed in 10% buffered formalin, embedded in paraffin, stained with hematoxylin and eosin, and blindly scored for hepatic lipid accumulation by microscopy examination by the same pathologist. The score of each component of NAFLD (steatosis [0–3], lobular inflammation [0–3], ballooning [0–2], and fibrosis [0, 1a, 1b, 1c, 2, 3]) was quantitatively evaluated according to the standard criteria of grading and staging for NASH [8].
Western blot
Hepatic tissues were isolated after pump-mediated perfusion of PBS, and whole cell lysates were prepared. Protein concentrations were determined and an equal amount of each sample was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Following the transfer, the membranes were reversibly stained with a Ponceau S solution (Amresco, Solon, OH, USA) to confirm sample loading and gel transfer equivalence. After blocking with 5% nonfat milk for 1 hour, the membranes were individually incubated with AMPK antibodies, followed by antibodies to phospho-AMPK (1:1,000; Cell Signaling, Danvers, MA, USA), SREBP-1c (1:1,000; BD Biosciences, San Jose, CA, USA), fatty acid synthase (FAS; 1:1,000; Cell Signaling), PPAR-α (1:1,000; Cell Signaling), CPT-1 (1:1,000; Cell Signaling), and actin (1:2,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. This was followed by incubation with goat-anti-rabbit immunoglobulin G secondary antibodies (1:2,000; Santa Cruz Biotechnology) for 1 hour. Antibody binding was detected by enhanced chemiluminescence (ECL, Pierce, Thermo Fisher Scientific, San Jose, CA, USA). X-ray film (Kodak, Rochester, NY, USA) was used to visualize the bands. Densitometry quantification values were measured by the Image J program from NIH and expressed as fold-differences from the control at each time point.
RNA isolation, semiquantitative reverse transcription-polymerase chain reaction (RT-PCR), and real-time RT-PCR
Total RNA was isolated from liver samples after pump-mediated perfusion with 5% PBS. The method was based on the phenol/guanidine isothiocyanate RNA zol B protocol (Cinna/Biotecx, Houston, TX, USA). Total RNA (1 μg) was reverse transcribed using MMLV (Moloney Murine Leukemia Virus) reverse transcriptase (Gibco, Grand Island, NJ, USA) with random hexamer priming. For semi-quantitative PCR, aliquots of cDNA were amplified in a 20-μL PCR mixture according to the protocol provided by the manufacturer (TaKaRa Bio, Kyoto, Japan). PCR primer sequences for genes regulating fatty acid synthesis and fatty acid oxidation are shown in Table 1. The primers were synthesized by Bioneer (Seoul, Korea). PCR products were separated by electrophoresis on 1.5% agarose gels containing ethidium bromide, and the bands were visualized under ultraviolet light. For real-time RT-PCR, the resultant cDNA was amplified using a LightCycler (Roche Diagnostics, Lewes, UK). Real-time PCR was carried out with SYBR Green I (Roche Diagnostics). PCR reactions with SYBR Green I and primers were performed in a 20 μL volume with 2 μL cDNA, 0.5 μM each primer, and 4 mM MgCl2. Taq polymerase, PCR buffer, deoxynucleotides, and SYBR Green I dye were included in the LightCycler-Fast Start DNA Master SYBR Green I mix (Roche Diagnostics). The thermal cycling profile consisted of a pre-incubation step at 95ºC for 10 minutes followed by 40 or 50 cycles of a 95ºC denaturation step for 10 seconds, 59ºC annealing for 5 seconds, and a 72ºC extension step for 20 seconds. After each extension step, the temperature was raised to 88ºC to measure SYBR Green I fluorescence at a temperature 2°C below the product Tm and 2°C above the Tm of the primers-dimers. At the end of the PCR, melting curve analysis was performed by gradually increasing the temperature from 65°C to 95°C at a rate of 0.1oC/second to confirm the amplification specificity of the PCR products. The level of expression of each mRNA and their estimated crossing points (Cp) for each sample were determined relative to the standard preparation using LightCycler computer software (version 3.5). PCR amplification was performed with a series of standards prepared by successive dilutions and a linear standard curve was automatically generated. A standard curve was constructed for each PCR run. All samples to be compared were run in the same assay.
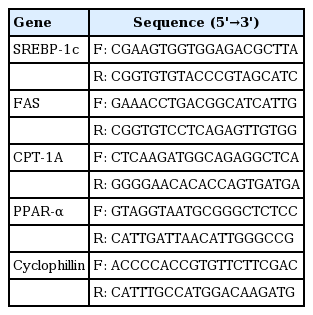
Primer sequences of genes related to fatty acid synthesis and fatty acid oxidation
Statistical analysis
The results are presented as the mean ± SD. The effect of CK on each parameter was examined by one-way analysis of variance. Prism software version 5 (Graphpad Software, San Diego, CA, USA) was used for statistical analysis and graphing. A p < 0.05 was considered statically significant.
RESULTS
Compound K improved glucose metabolism in HFD-fed OLETF rats
HFD-fed OLETF rats were treated for 12 weeks with saline (control), CK10, CK25, or metformin (as a positive control). The amount of food intake and the body weight were not significantly different among the four groups (Fig. 1A and 1B). Fasting plasma glucose levels of the CK25 and metformin groups were significantly lower than those of the control group during the 12-week study period (Fig. 1C). HbA1c was not significantly different among the four groups (Fig. 1D). Concerning OGTT, the glucose concentrations after glucose loading in the CK25 group were significantly lower than those of the control group (Fig. 2A). When the area under the curve (AUC) of glucose in the OGTT was analyzed, the AUC of the CK25 group was found to be significantly lower than that of the control group and decreased to the level of the metformin group (Fig. 2B). Fasting insulin levels of the CK25 and metformin groups were significantly lower than those of the control group were (Fig. 2C).
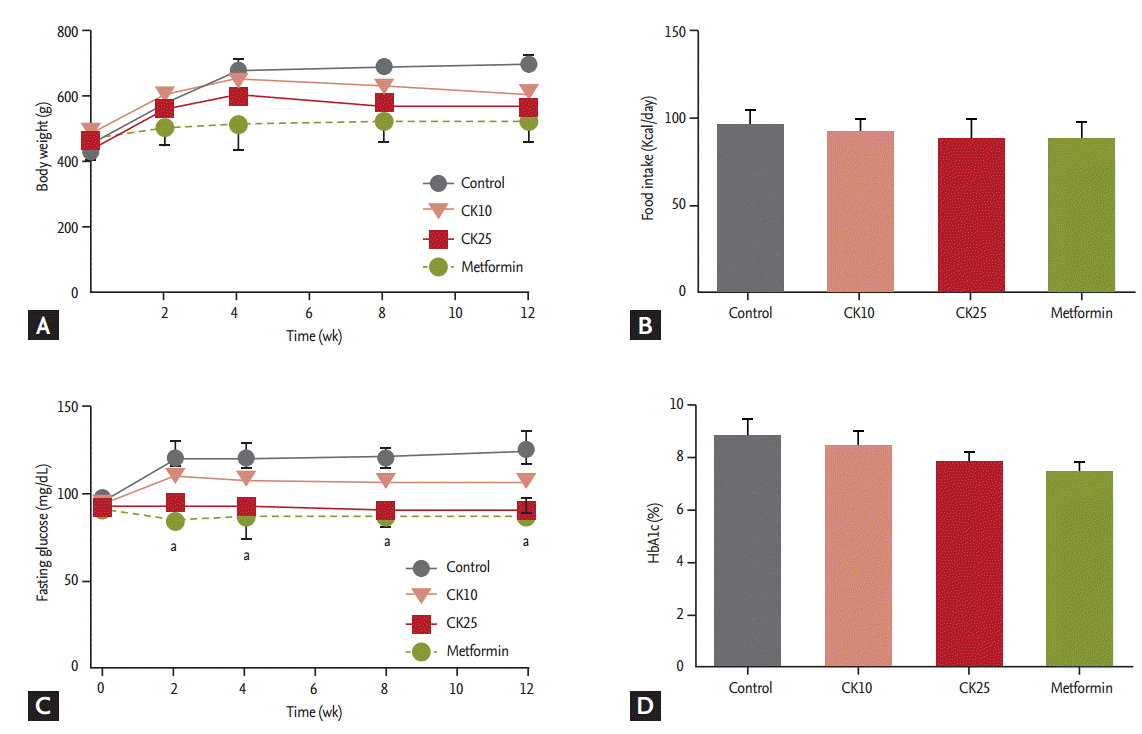
Effect of compound K (CK) supplementation on body weight, food intake, and fasting glucose concentrations in highfat diet-fed Otsuka Long-Evans Tokushima Fatty (OLETF) rats. Saline (control), CK 10 mg/kg, CK 25 mg/kg, or metformin 300 mg/kg was administered to OLETF rats for 12 weeks. (A) Body weight during 12 weeks. (B) Comparison of food intake among control, CK10, CK25, and metformin groups. (C). Serial fasting glucose levels for 12 weeks. (D) Glycated hemoglobin (HbA1c) levels. Data is expressed as mean ± standard deviation. aDifferences were considered statistically significant at p < 0.05.
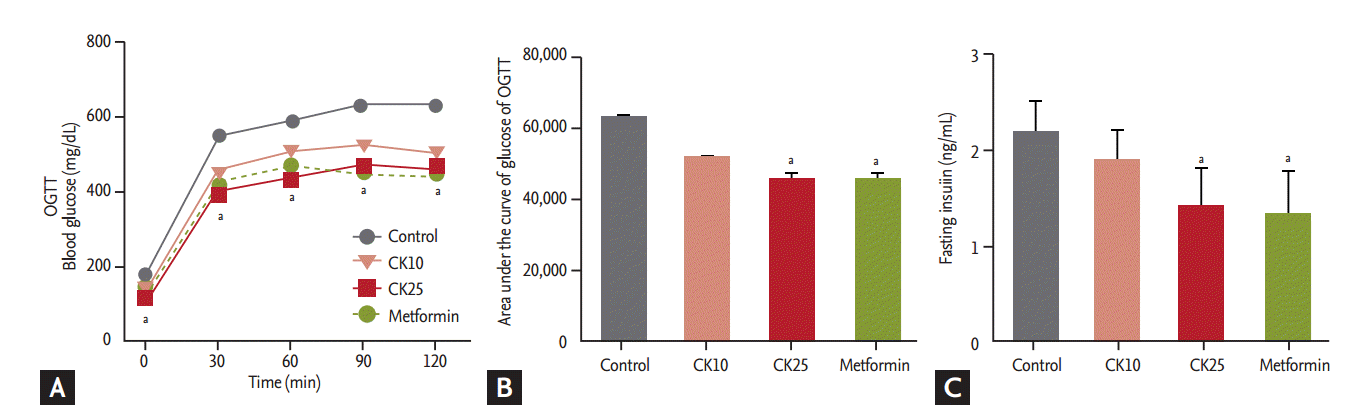
Effects of compound K (CK) in oral glucose tolerance tests (OGTT). OGTT was performed 12 weeks after treatment. (A) Blood glucose levels of OGTT. (B) Area under the curve of glucose levels of OGTT. (C) Fasting insulin levels measured at 12 weeks after treatment. Data is expressed as mean ± standard deviation. aDifferences were considered statistically significant at p < 0.05.
Compound K improved fatty liver in HFD-fed OLETF rats
To demonstrate the effect of CK on hepatic steatosis, lipid accumulation was measured in HFD-fed OLETF rats. Histological examination showed that hepatocytes of HFD-fed rats were distended by large cytoplasmic lipid droplets. The quantitative grade of hepatosteatosis was significantly lower in the CK25 group than in the control group (1.5 ± 0.82 vs. 2.7 ± 0.23, p < 0.05). Advanced fibrosis was not found in all groups. This change in cellular morphology was significantly improved by CK25 treatment (Fig. 3).
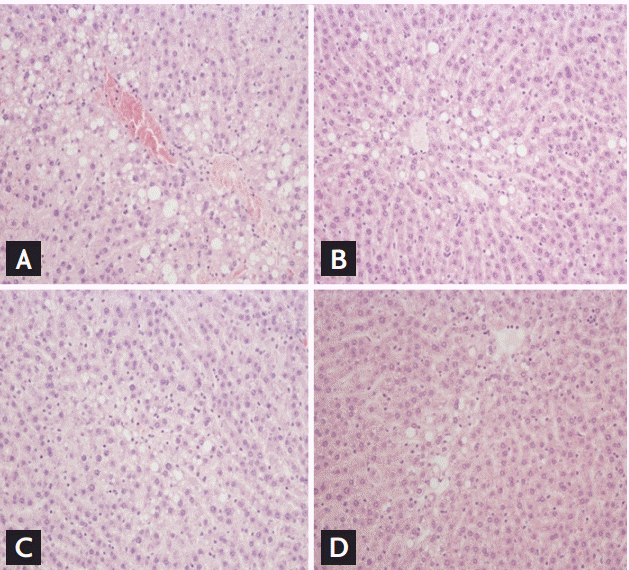
Effects of compound K (CK) on histological findings of hepatic steatosis. Liver tissue of (A) control, (B) CK10, (C) CK25, and (D) metformin groups were stained by H&E after 12 weeks of treatment (×100).
Compound K increased AMPK activation
To study the effect of CK on the phosphorylation of AMPK, which is a key regulator of glucose and lipid metabolism, the expression of phospho-AMPK in the liver of HFD-fed OLETF rats was examined by western blotting and compared among four groups. CK25 and metformin significantly increased hepatic AMPK phosphorylation (Fig. 4).
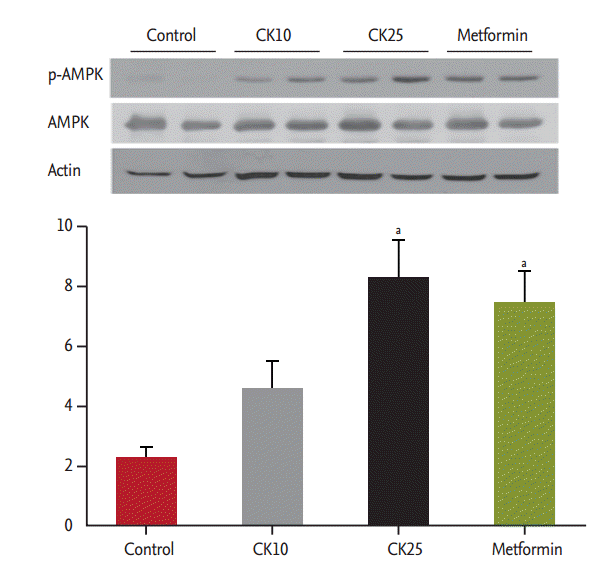
Effect of compound K (CK) on phosphorylation of adenosine monophosphate-activated protein kinase (p-AMPK). We compared the effect of CK on AMPK phosphorylation in the liver of high-fat diet-fed Otsuka Long-Evans Tokushima Fatty rats with that of the control group. CK25 and metformin significantly increased hepatic AMPK phosphorylation. aDifferences were considered statistically significant at p < 0.05.
Compound K decreased fatty acid synthesis and increased fatty acid oxidation
To investigate the mechanism by which CK improved hepatic steatosis in HFD-fed OLETF rats, the expression of genes related to fatty acid synthesis and β-oxidation was evaluated by real-time PCR and Western blotting. CK25 significantly inhibited the expression of SREBP-1c and FAS, which are involved in lipogenesis (Fig. 5). CK25 stimulated the mRNA expression of CPT-1, a rate-limiting enzyme for fatty acid oxidation (Fig. 5A). The mRNA expression of PPAR-α, a transcriptional factor that regulates fatty acid oxidation, was significantly increased by CK25 (Fig. 5A). In addition, western blotting showed that the expression of CPT-1 and PPAR-α in the CK25 and metformin groups were significantly higher than those of control groups (Fig. 5B).

Effects of compound K (CK) on lipogenic and lipolytic gene (A) mRNA expression and (B) protein expression in the liver of high-fat diet-fed Otsuka Long-Evans Tokushima Fatty rats. (A) Reverse transcription-polymerase chain reaction (PCR) was performed as described in methods. Transcripts of lipogenic and lipolytic genes were quantified by real-time PCR and normalized to cyclophillin. (B) Representative data of the Western blot showing the effect of CK on sterol regulatory element-binding protein-1c (SREBP-1c), fatty acid synthase (FAS), carnitine palmitoyltransferase-1 (CPT-1), and peroxisome proliferator-activated receptor-α (PPAR-α) expression (n = 5). Data is expressed as mean ± standard deviation. aDifferences were considered statistically significant at p < 0.05.
DISCUSSION
In the present study, CK attenuated glucose intolerance and hepatic steatosis in HFD-fed OLETF rats through the activation of AMPK. CK inhibited the expression of genes related to fatty acid synthesis and stimulated that of genes related to fatty acid oxidation through activation of the AMPK pathway. The histologic features of steatosis decreased in the CK25 treatment group.
NAFLD is a common typical feature of insulin resistance conditions such as obesity, type 2 diabetes, and metabolic syndrome [8]. Insulin resistance is associated with hyperinsulinemia, increases of plasma free fatty acid, and hepatic lipogenesis. Besides weight loss, multiple pharmacologic interventions have been attempted. Pentoxifilline [9], orlistat [10], ursodeoxycholic acid [11], metformin [12], and thiazolidinediones [13] have proved successful in humans. Insulin sensitizers such as pioglitazone [14] and metformin [15] improve hepatic steatosis by reducing hepatic insulin resistance through AMPK activation. Glucagon-like peptide-1 increases phosphorylated AMPK α1 (the unique subunit of AMPK) in human and rat hepatocytes [16]. Antioxidants including α lipoic acid [17] and vitamin E [18] also decrease hepatic lipogenesis through AMPK-dependent and -independent pathways.
CK is a metabolite of panaxadiol ginsenosides, which has antidiabetic and antihyperlipidemic activity. CK lowers blood glucose levels in db/db mice by stimulating insulin secretion and improving insulin resistance [19]. In the present study, CK decreased fasting glucose levels, the AUC of glucose in the OGTT, and the fasting insulin level. The results support a CK-mediated decrease in insulin resistance in HFD-fed OLETF rats. HbA1c levels decreased in the CK25 and metformin groups compared to that in the control group. A more definitive decrease could be apparent with a longer treatment period. In addition, we previously reported that CK reduces lipid accumulation through AMPK phosphorylation in HepG2 cells cultured in high-glucose medium [20]. In previous in vitro studies, CK activated AMPK phosphorylation and decreased gene expression of SREBP-1c and FAS; these effects were attenuated by compound C, which is an AMPK inhibitor. Thus, we hypothesize that CK inhibits hepatic steatosis by activating AMPK. In this study, we confirmed that CK attenuates glucose intolerance and hepatic steatosis in the liver of OLETF rats (an established type 2 diabetes model) through AMPK activation. In this model, hyperglycemia develops after 18 weeks of age [21]. An HFD- or methioninecholine-deficient diet is usually fed to induce steatohepatitis. We can study both glucose intolerance and hepatic steatosis in HFD-fed OLETF rats.
AMPK plays an important role in the regulation of cellular energy metabolism [22]. AMPK inhibits hepatic gluconeogenesis and reduces fatty acid synthesis by inhibiting the transcription factor SREBP-1c, and increases fatty acid β-oxidation through stimulation of CPT-1, which controls the transport of activated fatty acids into the mitochondria for oxidation [23].
SREBP-1 is a dominant transcription factor that regulates gene expression of lipogenic enzymes in the liver [24]. SREBP-1 is highly specific for lipogenic genes, whereas SREBP-2 is more specific to cholesterogenic genes. SREBP-1 has two isoforms. SREBP-1c is weaker than SREBP-1a and SREBP-2; the livers of adult animals predominantly synthesize SREBP-1c and SREBP-2 [25]. SREBP-1c over-expression in transgenic animals selectively activated fatty acid biosynthesis genes. In this study, CK inhibited the expression of SREBP-1c, suggesting that CK can also decrease the expression of lipogenic genes. SREBP-1c and its target proteins, such as FAS, were decreased by CK.
FAS is an important catalyzing enzyme in fatty acid biosynthesis [26]. FAS expression is regulated through the transcriptional factor SREBP-1c in the liver. In the present study, CK reduced hepatic FAS mRNA levels, which may contribute to the improvement of hepatic steatosis.
The regulation of lipid metabolism depends on the balance between biosynthesis pathways and hydrolysis. To improve fatty liver, both fatty acid oxidation and fatty acid synthesis are important. PPAR-α is a ligand-activated transcription factor that regulates the expression of genes related to fatty acid oxidation. PPAR-α promoter activity is increased in an AMPK-dependent manner [27]. PPAR-α transcriptionally regulates peroxisomal, microsomal, and certain mitochondrial fatty acid-metabolizing enzymes in the liver [28]. Herein, CK upregulated PPAR-α gene expression.
The mitochondrial carnitine system plays an important role in fatty acid oxidation by catalyzing fatty acid transport into the mitochondrial matrix during the catabolic (fasting) state. CPT-1 is one of the members of this transport system, which is localized in the mitochondrial outer membrane and regulated by changes in the malonyl-coenzyme A (CoA). The AMP-induced protein kinase primarily determines the concentration of malonyl-CoA [29]. CK activated AMPK phosphorylation, which decreased malonyl-CoA concentration and sequentially increased CPT-1 expression.
In conclusion, CK attenuated glucose intolerance and hepatic steatosis in HFD-fed OLETF rats through AMPK activation, which has a dual mode of action involving the decrease of fatty acid synthesis and the increase of fatty acid oxidation.
KEY MESSAGE
1. Non-alcoholic fatty liver disease is associated with insulin resistance.
2. Compound K (CK) is the final metabolite of panaxadiol ginsenosides that have been shown to exert antidiabetic effects.
3. CK improved glucose intolerance and hepatic steatosis through adenosine monophosphate-activated protein kinase activation.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This research was supported by a grant from Kyung Hee University in 2007 (KHU-20071482).