Apoptosis in Dilated Cardiomyopathy
Article information
Abstract
Objective
Cardiomyopathy, a popular diagnosis that always obscures more than it reveals, nevertheless has several characteristic histological features. These prominently include widespread focal myocardial fibrosis and associated hypertrophy of surviving cardiac myocyte, in fact, focal noninflammatory degeneration (not necrosis) has been demonstrated as a feature of many forms of cardiac hypertrophy. We hypothesized that this loss of myocardial cells in dilated cardiomyopathy (DCMP) may result from cell death by apoptosis.
Methods
Endomyocardial biopsy specimens from the right ventricles of six patients who suffered from DCMP were studied, and myocardial specimens from two persons who died in motor vehicle accidents were used as negative controls. For identification of apoptosis, immunohistochemistry with terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end-labeling was performed, in addition, apoptosis was confirmed morphologically by confocal laser scanning microscopy with propidium iodide.
Results
Apoptosis, that was represented by an apoptotic index ranging from 19.8 to 25.4%, could be extensively seen in myocytes and a/so rarely in non-myocytes of interstitium and vascular endothelium. Morphologically, there were a lot of nuclei with dumps of condensed chromatin, suggestive of apoptosis.
Conclusion
The present study demonstrated that myocyte loss in DCMP might be mainly due to the apoptosis of myocytes and interstitial cells, rather than inflammation or cell necrosis.
INTRODUCTION
Myocyte cell loss is a common characteristic of the failing heart. This phenomenon has been documented in humans and animals during the normal process of aging, following long-term pressure or volume overload hypertrophy, and in idiopathic DCMP1–6). Moreover, myocyte cell death is implicated in the architectural rearrangement occurring in the surviving myocardium, acutely and chronically after infarction7). However, until recently, the general belief was that cell necrosis was the exclusive mechanism of muscle cell death in the myocardium. In vivo and vitro studies have demonstrated that apoptosis is a significant form of myocyte death in the heart8–14).
Apoptosis, or programmed cell death, is a highly regulated and active process that contributes to the control of cell number during development and to the maintenance of many adult tissues15–17). It is triggered by the activation of an internally encoded suicide program as a result of either extrinsic or intrinsic signals18). The crucial role of apoptosis in pathologic conditions is increasingly being recognized19,20). Recently, apoptosis was reported as a possible mechanism for the loss of myocardial cells in an infant with Uhl’s anomaly21). Arrhythmogenic right ventricular dysplasia, a form of right ventricular cardiomyopathy that commonly leads to severe ventricular arrhythmias and sudden death, is characterized by noninflammatory loss of myocardial cells and their progressive replacement by fat and fibrous tissue22–24). These findings indicate that apoptotic myocardial cell death occurs and may contribute to the loss of myocardial cells in this disorder25). Moreover, apoptosis contributes to ventricular remodeling in pacing-induced heart failure in dogs and is the prevailing type of myocardial damage associated with myocardial ischemia and reperfusion in rabbits and rats11,12,26).
We hypothesized that loss of myocardial cells in DCMP may result from cell death by apoptosis, and this phenomenon may be of high magnitude in the loss of myocytes compared with other studies6,12,14,25,27). For identification of apoptosis, immunohistochemistry with terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end-labeling (TUNEL) was performed. In addition, apoptotic nuclear change was confirmed by confocal laser scanning microscopy with propidium iodide. The present study demonstrated that the ongoing myocyte loss in DCMP might be mainly due to the apoptosis of myocytes and interstitial cells, rather than inflammation or cell necrosis.
MATERIALS AND METHODS
1. Cardiac Specimens
Endomyocardial biopsies were done at the right ventricle of six patients (five men and one women; mean age 46.2 years, three specimens for each patient, n = 18) who suffered from mild and moderate dyspnea on exertion (New York Heart Association [NYHA] classification II or III). Transthoracic echocardiography of all patients suggested global left ventricular dysfunction (mean left ventricular ejection fraction [LVEF] 31±4%) and eccentric hypertrophy associated with atrioventricular valvular regurgitation. To exclude coronary artery diseases, coronary angiography and left ventricular angiography were performed in all patients. Two patients had minimal coronary artery disease and four patients had normal coronary arteries. There were no regional wall motion abnormalities in left ventricular angiography, except global left ventricular dysfunction (Table 1). The pathologic diagnosis of DCMP was based on the following established criteria: interstitial fibrosis and fat tissue deposition with surviving strands of cardiomyoctes embedded in or bordered by fibrous tissue, a typical finding of the patchy replacement of myocardium found in mild to moderate interstitial fibrosis, and fatty tissue infiltration associated with hypertrophic change of myocytes28–30). These criteria were present in all sections of the patients studied. Myocardial samples from two persons who died in motor vehicle accidents were used as negative controls (three specimens for each patient, n = 6).
Clinical and hemodynamic characteristics of the six DCMP patients before medical therapy endomyocardial biopsy
Tissues were fixed in 10 percent buffered formalin and embedded in paraffin. Four to six sections (5m) from each paraffin block were analyzed for the presence of apoptosis.
2. Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end-labeling assay
As a result of endonulease activity, genomic DNA strand fragments are commonly formed during the apoptotic process within the cell nuclei. The TdT-mediated dUTP nick end-labeling (TUNEL) methods labels the ends of DNA fragments and is, therefore, used to detect apoptotic cells in situ. In the present study, identification of apoptosis in myocardial cross-sections was performed by TUNEL technique31), using Boehringer Mannheim In-situ Cell Death Detection Kit (Boehringer Mannheim, Indianapolis, IN) with a modified protocol. Briefly, cross-sections were deparaffinized and rehydrated through the following solution: xylene twice for five minutes, 100% ethanol twice for two minutes, 95% ethanol twice for two minutes and phosphate buffered saline (138 mM NaCl and 2.7 mM KCI; Sigma, St. Louis, MO). Tissues were treated with 25 μg/ml proteinase K (Boehringer Mannheim, Indianapolis, IN) for 20 minutes at 37°C to strip the proteins of nuclei, and then rinsed again with phosphate buffer solution. The TUNEL reaction mixture consisting of terminal deoxynucleotidyl transferase from calf thymus in storage buffer and nucleotide mixture in reaction buffer, and normal swine serum (Vector Laboratories, Burlingame, CA) were applied to the slides, coverslipped and incubated in humidity trays at 37°C for an hour. The slides were rinsed in phosphate buffer solution and sheep anti-fluorescein antibody conjugated with alkaline phosphatase (diluted 1:5 in 100 mM tris-HCI, and 150 mM NaCl at pH=7.5) was applied to the slides and incubated at 37°C for 30 minutes and rinsed again in phosphate buffer solution. The slides were then washed in buffer C (100 mM NaCl, 100 mM tris-base, 14.85 mM magnesium chloride at pH=9.5). Nitroblue tetrazolium chloride 5-bromo-4-chloro-3-indolyphosphate p-toludine salt color development (Gibco BRL, Gaithersburg, MD) was performed for 12 minutes, maximal. The slides were counterstained with Nuclear Fast Red (Sigma, St. Louis, MO) or methylene blue, dehydrated and coverslipped.
All slides were reviewed by at least two investigators. Apoptotic nuclei were identified by the presence of dark blue staining. For each specimen (three specimens for each patients, n=18), tissue sections were examined microscopically at 40X magnification and at least 200 cells were counted in five high-power fields. The degree of apoptosis was determined by the apoptotic index (%); numbers of (+) stained myocyte nuclei 100/total numbers of myocyte nuclei counted. Average mean apoptotic index for each patient was calculated. Stained cells at the edges of tissues were not counted because of inadequate tissue condition for counting. An apoptotic index of 2 or less was considered to indicate the absence of apoptosis. Myocardial samples from two persons who died in motor vehicle accidents were used as negative controls (three specimens for each patients, n=6). Positive control, which was purchased from Oncor (Gaithersburg, MD), consisted of rodent mammary glands obtained on the fourth day after weaning of rat pups.
3. Immunohistochemical Detection of Caspase-3
Caspase-3 (CPP-32) is a cysteine protease required for the initiation of apoptotic cell death32). It is related to interleukin-1 converting enzyme (ICE) and CED-3, the product of a gene required for programmed cell death in the nematode Caenorhabditis elegans. Caspase-3 is the specific ICE/CED-3-like mammalian cysteine protease that cleaves and inactivates poly (adenosine diphosphate ribose) polymerase (PARP), and enzyme involved in DNA repair and genome integrity, and thus may be the human equivalent of CED-3. Therefore, to provide further evidence of the occurrence of apoptosis in DCMP, we analyzed the level of expression of caspase-3 in the sections of the patients and controls using immunohistochemical techniques.
After deparaffinization and rehydration, the sections were incubated with 1:10 normal horse serum for 30 minutes at room temperature, washed once in phosphate-buffered saline, and incubated with a mouse monoclonal anti-CPP-32 antibody (Trans-duction Laboratories, Lexington, KY) at dilution of 1:1000. The slides were biotinylated with horse antimouse IgG (Vector Laboratories, Burlingame, CA) at a dilution of 1:200. Color reaction was visualized with an avidin-alkaline phosphatase-substrate system (Vectastain ABC Kit and Vector Red, Vector Laboratories, Burlingame, CA). As a negative control, adjacent sections were stained without the primary antibody against CPP-32.
4. Confocal laser scanning microscopy - propidium iodide (PI) assay
Apoptosis has been defined by a number of ultrastructural criteria, including cell size and gross structure, membrane permeability and integrity, as well as chromatin and DNA structure15,33,34) To evaluate nuclear chromatin morphology, fluorescent DNA-binding propidium iodide (PI) stain was done under confocal laser scanning microscopy.
Immediately after biopsy from the heart, myocardial specimens were subsequently placed in 3 ml of DME-H21 culture media (Gibco BRL, Gaithersburg, MD) with 20% fetal bovine serum (FBS, Gibco BRL), penicillin (100 U/ml), streptomycin (50mg/ml) and fungizone (25mg/ml), and incubated in humidified atmosphere containing 8% CO2 at 37°C. After equilibration to this temperature, myocardial specimens were labeled with 5μg/ml of PI (Molecular Probes, Eugene, OR) for 30 minutes. In a preliminary experiments, the incubation periods of PI, ranging from 10 to 40 minutes, were evaluated; an incubation period of 30 minutes allowed the best detection of increased PI staining. After the incubation, myocardial specimens were washed three times with phosphate buffered saline for five minutes, each, cut and placed under a confocal laser scanning microscopy on a slide glass. This form of confocal microgram is based on the optical sectioning abilities of the confocal microgram to obtain en face sequential images of cells at depth up to 1 μm. Condensed and/or fragmented apoptotic nuclei were identified by the presence of bright red fluorescence.
Experiments were performed in duplicate and analyzed on a confocal laser scanning microscopy LSM 310 (Carl Zeiss Inc., Germany) equipped with one argon/krypton laser. PI excitation took place on an excitation wave length of 488nm, and emission was collected through 630/22 and 530/30 nm band-pass filters, respectively. Images were acquired with a 40 neofluor lens with 1.3 n.a., digitized with a matrix of 512×512 pixels with a resolution of 0.3125 μm per pixel and analyzed by using the LSM 310 software on an IBM work station.
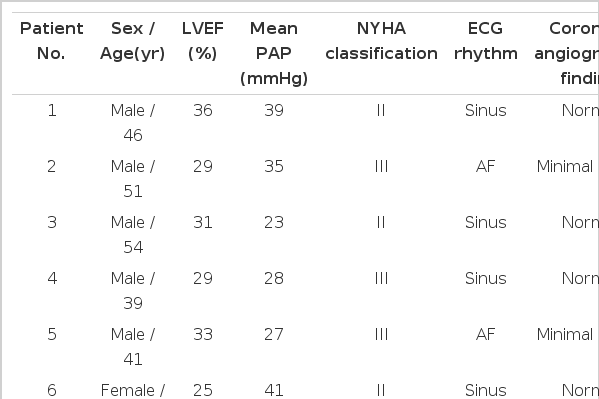
Confocal microscopy was also utilized for the identification of myocyte pattern in the hematoxylin-eosin (H&E) staining of myocardial specimens. The histologic architectures and elastin patterns of H&E-stained coronary arteries under light microscopy were also compared with those under the confocal laser scanning microscopy, using corresponding sections35) (Figure 1). All the experiments were performed in duplicate and analyzed on a confocal laser scanning microgram LMS310 (Carl Zeiss Inc., Germany) equipped with an argon/krypton laser. The myocardial cells were visible on an excitation wavelength of 488/568 nm and the emissions collected through a 530/20 nm band-pass filter (pinhole=20). Images per specimen were examined using a 10 neofluor lens with 0.3 n.a., digitized with a matrix of 512×512 pixels with a resolution of 0.625 m per pixel and a field size of 0.1024mm2.
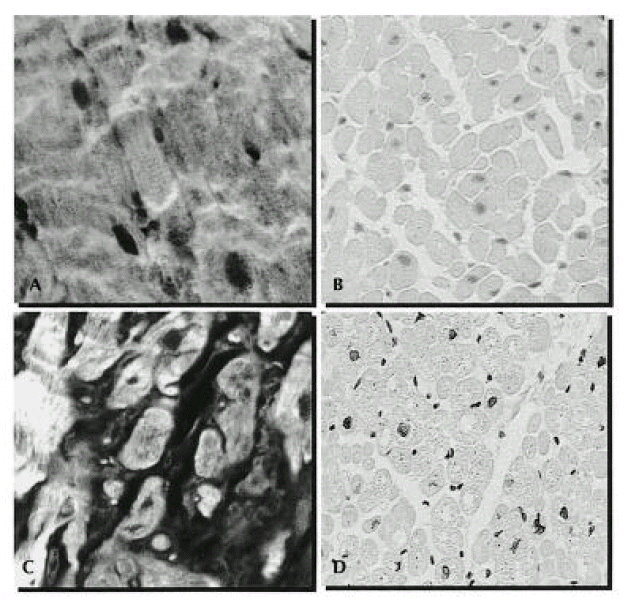
Confocal laser micrograms of myocardium from a non-cardiac death (Panel A) and a patient with DCMP (Panel C, magnification ×1280). Compared with normal myocardium, transverse section of myocardium of DCMP reveal disarray and hypertrophic change of myocytes with interstitial fibrous septa. In panel B, a cross-section of myocardium from a non-cardiac death reveals no apoptosis. Extensive apoptosis (black nuclei) can be seen in myocyte and also observed in rare interstitial cells in panel D (TUNEL for apoptotic nuclei and methylene blue counterstaining, ×40).
5. Statistical analysis
Differences between apoptotic index of patients with DCMP and negative control were assessed by a one-way ANOVA test followed by Mann-Whitney U-Wilcoxon Rank Sum W test. A value of p < 0.05 was considered significant in all analyses. All data in the text and figures are presented as mean ± SEM
RESULTS
Myocardial specimens from two persons who died in motor vehicle accidents showed a normal pattern of myocytes (Figure 1A); blood vessels and interstitial cells in the myocardium were normal. Compared with normal myocardium, histologic findings from the myocardium of DCMP showed hypertrophic changes and loss of the longitudinal arrangement of myocytes with mild-to-moderate interstitial fibrosis, but inflammatory infiltrates were absent (Figure 1C and 2A). Confocal laser scanning micrography of the corresponding section showed disarray and hypertrophic change of myocytes with vacuolization of cytoplasm (Figure 1C).
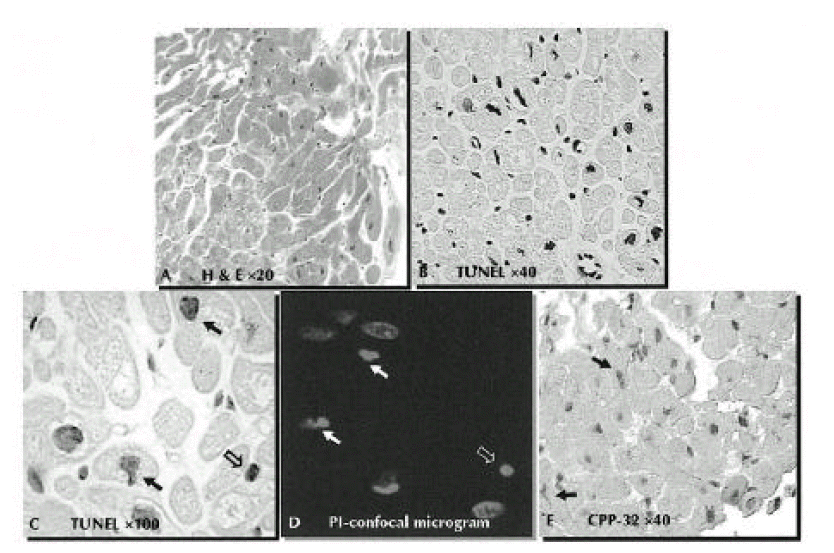
Evidence of apoptosis in dilated cardiomyopathy. In panel A, a myocardial section from a patient with DCMP (Patient 6) shows hypertrophic changes of myocytes with vacuolization and interstitial fibrosis (H&E staining, × 20). There was no inflammatory cell infiltration. Extensive apoptosis can be seen in the myocyte, but apoptosis is rarely observed in the non-myocytes of interstitium and vascular endothelium in panel B (TUNEL for apoptotic nuclei and methylene blue counterstaining, ×40). In panel C, microscopic morphology of nucleus shows chromatin condensation (open arrow) and small clump of condensed chromatin (filled arrows) (TUNEL for apoptotic nuclei and methylene blue counterstaining, ×100). Confocal laser scanning micrography using a nucleus labeling with propidium iodide (Panel D). There are small, homogenous, condensed nuclei, suggestive of apoptosis (×1280, open arrow). In the same field, residual apoptotic bodies, appearing as small clumps of condensed chromatin, are evident (filled arrows). Immunohistochemical detection of caspase-3(CPP-32) (Panel E, ×40). The antibody for CPP-32 is detected with anti-mouse IgG conjugated with biotin and an avidin-alkaline phosphatase-substrate system in which positive cells are stained brown (filled arrows).
By TUNEL stain, apoptosis could be detected in myocardial specimens from all six patients with dilated cardiomyopathies (Figure 1D, 2B and 2C). The majority of these TUNEL(+) cells were myocytes which were easily recognized under a light microscope at high magnification, but apoptosis was rarely observed in non-myocytes of the interstitium and vascular endothelium (Figure 2B). The apoptotic index ranged from 19.8 to 25.4% (Table 2). Control samples from two patients who died in motor vehicle accidents were negative in TUNEL stain.

Evidence of apoptosis in situ end-labeling
To eliminate the possibility of labeling of nonapoptotic nuclei and the potential overestimation of this form of myocyte cell death, and to evaluate the morphology of the nuclear chromatin, confocal laser micrography with PI was compared with corresponding sections of TUNEL stain. Figure 2D shows morphological alterations in chromatin structure of myocytes; margination of condensed chromatin and small clumps of condensed chromatin are suggestive findings of apoptotic bodies.
Caspase-3 is important for the induction of apoptotic cell death in mammalian cells. It was also detectable in myocardial specimens of all six patients with DCMP with immunohistochemistry (Figure 2E). In normal controls, caspase-3 could not be detected.
DISCUSSION
In the present study, apoptosis was identified by in situ end-labeling of fragmented DNA with TdT and biotinylated dUTP, a commonly accepted method for the detection of the apoptotic process36). Apoptotic cells were observed mainly in myocytes, but rarely in interstitial cells. To correlate chromatin alteration with the presence of dUTP labeling, different regions of myocardium were analyzed by confocal laser scanning microscopy with PI. There were some nuclei with clumps of condensed chromatin, suggestive of apoptosis. In the same field, residual apoptotic bodies, appearing as small clumps of condensed chromatin, were evident. The inexorable decline in cardiac function seen in DCMP despite the absence of an active inflammatory process might be partially explained by apoptosis. Comparing with other study6,25), the present result of apoptotic index using a dUTP labeling was similar and also discrepancy of the magnitude of this phenomenon obtained by the TdT reaction with a fluorescence probe27). To gain further evidence of apoptosis in DCMP and extend our findings, we examined the expression of protease caspase-3, which is responsible for the cleavage of PARP. Our finding that high levels of caspase-3 expression were associated with positive in situ labeling of fragmented DNA provides strong evidence of apoptotic cell death in DCMP. The exact incidence of apoptosis in heart failure has been debated and it is likely that conflicting degrees of apoptosis are dependent on the method used for detection and may also be dependent on the method of tissue preservation prior to tissue evaluation37).
The factors that lead to the development and progression of heart failure are still not fully understood, but increased apoptosis has been proposed as one mechanism38). Recent studies have demonstrated that apoptosis in human cardiac myocytes is significantly increased in end-stage heart failure at the time of transplantation or at autopsy6,27). This increased apoptosis is present in both idiopathic dilated cardiomyopathy and ischemic cardiomyopathy and has been confirmed by histochemical evidence of DNA fragmentation and DNA laddering. This abnormal regulation of myocyte cell death has lead to many theories of the mechanism and potential pathogenesis of progressive heart failure. A number of abnormalities found in heart failure, including increased myocyte cytosolic free calcium, hypoxia, increased superoxide radicals, and increased muscle stretch, have all been shown to increase apoptosis14,39). In addition, increased apoptosis may be a manifestation of the excessive growth or maladaptive hypertrophy of terminally differentiated cardiac myocytes in heart failure40).
Recent reports of molecular abnormalities among patients with cardiomyopathy support that apoptosis might be responsible for cardiomyopathy6,41,42). There could be apoptotic cell death of selected but spatially distributed cardiac myocytes. Extracellular ATP, as may occur after any death of a cardiac myocyte, may itself trigger apoptosis and establish a vicious cycle of cellular destruction43). There is compelling evidence that viral myocarditis is very often a functionally important participant in the pathogenesis of cardiomyopathy44,45). This could represent independent coexistence with apoptosis, but there are new and intriguing examples in which several different viruses acted as triggers of apoptosis46–50). This possibility in relation to the development of cardiomyopathy could explain many of the otherwise puzzling ways by which viral myocarditis does more than simply direct viral destruction of myocytes. In this study, notably, infiltration of inflammatory cells was absent in the sections from patients with DMCP and is coincides with other studies. This suggests that cell-mediated cytotoxicity by immune cells appears not to play a major role under these conditions51).
Although there is general consensus that segmental and replacement fibrosis are the con sequence of the reparative processes following myocyte cell loss, the cause of interstitial fibrosis remains to be defined. The problem concerns whether this type of interstitial alteration occurs through activation of fibroblasts via humoral or mechanical factors in the absence of myocyte loss, or whether cell death is required for the stimulation of the growth response of the nonmyocyte compartment of the myocardium52,53). The observation that apoptosis is present in various pathologic states of the heart raises questions about the mechanism responsible for modification of the interstitium with accumulation of fibrillar collagen. Death of individual myocytes may be more common than generally believed, as with our results, and this phenomenon may stimulate discrete healing processes contributing to the expansion of the interstitium54). Myocytolytic necrosis and apoptosis may coexist not only in the development of segmental and replacement fibrosis, but also in the formation of interstitial fibrosis55,56). Importantly, programmed cell death is not restricted to myocytes and affects interstitial fibroblasts as well8,9,14). Fibroblast apoptosis may have a role as a possible mechanism for fibrosis in heart failure53). Finally, interstitial fibrosis reflects widening of the extracellular space with collagen deposition between groups of myocytes as a result of diffuse myocyte cell deaths in the wall7).
In summary, our results support the hypothesis that the loss of myocardial cells, including myocytes and interstitial cells in DCMP, may result from cell death by apoptosis rather than inflammation or cell necrosis. At present, although the contribution of apoptosis to progressive heart failure is unknown, it seems possible that this phenomenon plays a significant role; further investigation and therapeutic trials can be expected in the future.