Insulin resistance is a major risk factor for developing type 2 diabetes caused by the inability of insulin-target tissues to respond properly to insulin, and contributes to the morbidity of obesity. Insulin action involves a series of signaling cascades initiated by insulin binding to its receptor, eliciting receptor autophosphorylation and activation of the receptor tyrosine kinase, resulting in tyrosine phosphorylation of insulin receptor substrates (IRSs). Phosphorylation of IRSs leads to activation of phosphatidylinositol 3-kinase (PI3K) and, subsequently, to activation of Akt and its downstream mediator AS160, all of which are important steps for stimulating glucose transport induced by insulin. Although the mechanisms underlying insulin resistance are not completely understood in skeletal muscle, it is thought to result, at least in part, from impaired insulin-dependent PI3K activation and downstream signaling. This review focuses on the molecular basis of skeletal muscle insulin resistance in obesity and type 2 diabetes. In addition, the effects of insulin-sensitizing agent treatment and lifestyle intervention of human insulin-resistant subjects on insulin signaling cascade are discussed. Furthermore, the role of Rho-kinase, a newly identified regulator of insulin action in insulin control of metabolism, is addressed.
A fundamental mechanism for the maintenance of glucose homeostasis is the rapid action of insulin to stimulate glucose uptake and metabolism in peripheral tissues. Skeletal muscle is the primary site of glucose disposal in the insulin-stimulated state [1]. Resistance to the actions of insulin in skeletal muscle is a major pathogenic factor in type 2 or type 1 diabetes mellitus [2,3]; it also contributes to the morbidity of obesity, and complicates poorly controlled type 1 (autoimmune) diabetes [2]. The ability of insulin to increase glucose transport in skeletal muscle is elicited by the translocation of glucose transporter 4 (Glut4), the major insulin regulated glucose transporter, from intracellular vesicles to the plasma membrane and transverse tubules [4]. In muscle of type 2 diabetic subjects, the expression of the Glut4 gene is normal, and impaired glucose uptake by insulin action most likely results from altered trafficking or impaired function of Glut4 [5-7]. Because glucose transport in response to other stimuli that use different signaling pathways is normal in muscle of type 2 diabetic subjects [4], the resistance to insulin stimulation may be due to impaired insulin signal transduction [8]. In this review, we mainly summarize the updated information on insulin signaling over the past decade, with particular emphasis on the molecular mechanism of human insulin resistance, and also address the physiological role of the newly identified player of insulin action.
Insulin receptor signaling
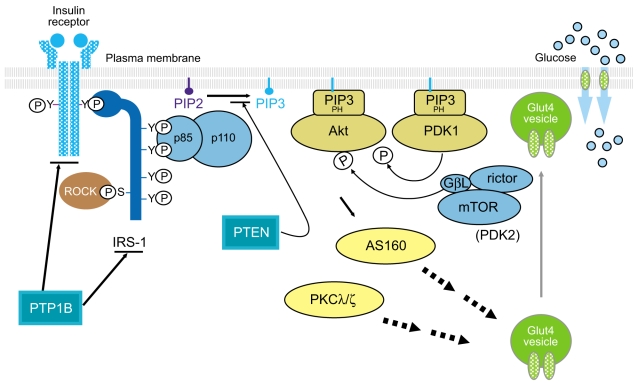
Insulin signaling involves a cascade of events initiated by insulin binding to its cell surface receptor, followed by receptor autophosphorylation, and activation of receptor tyrosine kinases, which result in tyrosine phosphorylation of insulin receptor substrates (IRSs) including IRS1, IRS2, IRS3, IRS4, Gab1, and Shc [9,10]. Binding of IRSs to the regulatory subunit of phosphoinositide 3-kinase (PI3K) via Src homology 2 (SH2) domains results in activation of PI3K, which phosphorylates membrane phospholipids and phosphatidylinositol 4,5-bisphosphate (PIP2) on the 3' position. This complex activates the 3-phosphoinositide-dependent protein kinases (PDK-1 and PDK-2) resulting in activation of Akt/protein kinase B (PKB) and atypical protein kinase C λ and ζ, (PKCλ/ζ), each of which are serine/threonine kinases [11,12]. Activated Akt phosphorylates its 160 kDa substrate (AS160), which stimulates the translocation of insulin-mediated Glut4 from intracellular vesicles to the plasma membrane [13]. Moreover, activation of PKCλ/ζ is also involved in the regulation of Glut4 translocation in response to insulin [14,15]. However, the insulin receptor (IR) is also dephosphorylated and inactivated by protein tyrosine phosphatases (PTPs), which comprise an extensive family of proteins that exert negative effects on insulin action and glucose metabolism [16,17]. In addition, phosphatase and tension homologue deleted on chromosome 10 (PTEN), a lipid phosphatase, serves as an important negative modulator for the insulin signaling pathway by hydrolyzing phosphatidylinositol 3,4,5-triphosphate to PIP2, antagonizing the PI3K pathway [18,19]. Thus, the physiological regulation of insulin action is controlled by the balance between phosphorylation and dephosphorylation (Fig. 1). Most importantly, the PI3K pathway is thought to be a key component of the insulin signaling cascade, which is necessary for the metabolic effects of insulin on glucose transport and Glu4 translocation [20,21]. Indeed, insulin-stimulated PI3K activity decreases in skeletal muscle of type 2 diabetic subjects [8,22], providing evidence for a defect in insulin signaling that could contribute to impaired Glut4 translocation and insulin resistance.
Role of Akt in insulin signaling
The serine/threonine kinase Akt is a downstream mediator of PI3K [12]. Three Akt isoforms have been cloned [23,24]; Akt1, Akt2, and Akt3, all of which are ubiquitously expressed in the tissues. Insulin has differential effects on Akt isoforms in a tissue-, isoform-, and species-specific manner [25,26]. In obese rats, insulin-stimulated Akt1 activity decreases in muscle and adipose tissue but increases in liver, while insulin-stimulated Akt2 activity decreases in muscle and liver but increases in adipose tissue [25]. Some studies have shown that inhibition of Akt activation by DN-Akt expression has no significant effect on insulin-stimulated glucose transport, whereas Akt siRNA inhibits insulin-stimulated glucose transport in 3T3-L1 adipocytes [27]. Furthermore, in skeletal muscles from insulin-resistant models, including PTP-1B transgenic mice [28], leukocyte antigen-related phosphatase (LAR) transgenic mice [29], and glucosamine-infused rats [30], insulin-induced Akt activation is normal but insulin-stimulated glucose uptake is impaired. In skeletal muscle of obese insulin resistant humans with or without type 2 diabetes, the effect of insulin on the activity of all Akt isoforms in muscle in vivo is normal [8]. However, at very high insulin levels in vitro, Akt activity is diminished in muscle from non-obese type 2 diabetics [31]. Moreover, Akt2 phosphorylation is impaired in adipocytes from obese type 2 diabetics [32]. Whether these impairments in Akt1 or Akt2 activity are sufficient to play a role in insulin resistant states is unclear. Akt1 knockout mice do not have insulin resistance, although their growth retardation or developmental effects could mask this [33]. Akt2 knockout mice have impaired insulin action on liver and modest effects in muscle and adipocytes that could possibly be secondary to the liver defect [34]. In this regard, our studies have also demonstrated that insulin-stimulated Akt activity in skeletal muscle of these obese rats is very mildly reduced while PI3K activity is markedly reduced, as compared to lean rats [25]. These data lead us to propose the hypothesis that full activation of PI3K is not necessary to maximally activate Akt, and that other pathways that are independent of PI3K are also involved in activation of Akt by insulin, at least in insulin resistant states. Data in support of the hypothesis come from studies showing that insulin-stimulated PI3K activation is reduced by 50% to 60% while Akt activation is normal in hepatoma cells transduced with a dominant-interfering mutant of dynamin [35]. Conceivably, such pathways could be induced in altered metabolic states such as obesity or type 2 diabetes. Thus, the potential impact of altered Akt activity in insulin-dependent glucose metabolism remains uncertain. This important issue can be resolved by studying an animal model that has Akt selectively deleted in peripheral tissues. Until now, there are no data regarding the effects of tissue-specific deletion of Akt on glucose metabolism and insulin signaling.
AS160 is an Akt substrate involved in Glut4 translocation
Investigation of steps down-stream of Akt could shed light on the role of Akt in insulin resistance. Akt directly phosphorylates cytosol proteins that have the RXRXXS/T motif. Using a phosphospecific antibody that only recognizes serine or threonine residues, which are phosphorylated by Akt [36], the Lienhard group has discovered the AS160 from 3T3-L1 adipocytes. This molecule was originally named TBC1 domain family member 4 (TBC1D4) and contains two phosphotyrosine binding (PTB) domains at the NH2 terminus and a Rab GTPase activating protein (GAP) domain at the COOH terminus [37]. In the basal state of 3T3-L1 adipocytes, AS160 was mainly localized in the low density microsomes (LDM) fraction. However, upon insulin stimulation, it was redistributed from the LDM compartment to the cytosol, but these effects were significantly blocked by treatment with a PI3K inhibitor [36]. Given that one of the LDM components are vesicles containing the glucose transporter Glut4 [38], and they move to and fuse with the plasma membrane in response to insulin [39], it is conceivable that AS160 may be involved in the process of GLUT4 trafficking that is dependent on PI3K signaling. The Lienhard group further demonstrated the important role of AS160 in the regulation of Glut4 translocation by showing that insulin causes a marked increase in AS160 phosphorylation at Ser318, Ser570, Ser588, Thr642, and Thr751 residues, and that mutation of these sites inhibits Glut4 translocation in response to insulin [13]. Support for this finding comes from skeletal muscle studies showing that insulin stimulates AS160 phosphorylation in skeletal muscle in a PI3K-dependent fashion, and that contraction and the AMP-activated protein kinase (AMPK) activator aminoimidazole carboxamide ribonucleotide (AICAR) also increases AS160 phosphorylation in isolated rat epitrochlearis muscle and other muscle systems [40-44]. Moreover, in skeletal muscle of human insulin-resistant subjects, including those with polycystic ovary syndrome and type 2 diabetes, the ability of insulin to increase AS160 phosphorylation is significantly impaired [45,46]. Collectively, the current available data provide important evidence that insulin-stimulated phosphorylation of AS160 is required to regulate Glut4 translocation, which is a critical step in controlling glucose homeostasis, and that decreased insulin-induced AS160 phosphorylation in skeletal muscle may play an important role in insulin resistance in vivo.
Role of PKCλ/ζ in the regulation of insulin-mediated glucose transport
The atypical PKC isoforms λ and ζ, are downstream PI3K mediators, and their activation is required for insulin stimulation of glucose uptake [47-49]. Overexpression of a dominant negative mutant of PKCλ or PKCζ abrogates insulin-stimulated glucose transport and Glut4 translocation in adipose [47,50] and muscle cells [48,49]. Overexpression of constitutively active PKCλ in adipocytes [47] or wildtype PKCζ in muscle in vivo [51] enhances both basal and insulin-stimulated glucose transport. In addition, PKCλ and ζ appear to function interchangeably, as overexpression of wild type PKCλ restores the inhibitory effects of a dominant negative mutant of PKCζ on insulin-stimulated Glut4 translocation, and vice versa [52]. The possibility that PKCλ/ζ could play an important role in insulin resistance in vivo is supported by studies showing impaired insulin-stimulated PKCλ/ζ activity in skeletal muscle and adipose tissue of non-obese type 2 diabetic GK rats [53,54]. Moreover, insulin-stimulated PKCλ/ζ activity decreases in cultured myotubes of obese humans with impaired glucose tolerance [55] and in muscle of obese diabetic monkeys [56]. Along with these findings, the Farese group have demonstrated that muscle-specific deletion of PKCλ/ζ causes insulin resistance by reducing insulin-mediated glucose transport in skeletal muscle [15]. Importantly, insulin-stimulated PKCλ/ζ activity is reduced in the muscle of obese nondiabetic and obese type 2 diabetic subjects. This contrasts with our findings that Akt activation is normal in obese and diabetic subjects with similar metabolic characteristics [8]. Thus, although insulin-induced PI3K activity is reduced in type 2 diabetic subjects, not all downstream pathways are similarly affected. These data imply that reduced insulin-stimulated atypical PKC activity may play an important role in insulin resistance in vivo.
Insulin-sensitizer agents and insulin signaling
Thiazolidinediones (TZDs) are a new class of insulin-sensitizing agents being used for the treatment of type 2 diabetes [57]. The molecular targets of these compounds are thought to include the nuclear receptor, peroxisome proliferator activator receptor-γ (PPARγ), which regulates the expression of numerous genes that affect glucose and lipid metabolism [58]. Evidence suggests that TZDs ameliorate insulin resistance in humans primarily by increasing insulin-stimulated glucose disposal in skeletal muscle [59]. Our previous studies revealed that treatment with troglitazone, a member of the TZD family, increases insulin-stimulated IRS-1-associated PI3K activity and Akt activity in skeletal muscle of type 2 diabetic patients [60]. The troglitazone effect on PI3K activity was associated with an increase in the amount of the p110β catalytic subunit of PI3K [60]. Consistently, enhanced Akt phosphorylation was also detected in skeletal muscle in normal, glucose-tolerant, insulin-resistant, first-degree relatives of type 2 diabetic patients [61]. These findings suggest that the mechanism for the insulin-sensitizing effect of TZDs could involve enhanced PI3K activation in skeletal muscle of obese type 2 diabetic subjects. However, Karsson et al. [62] demonstrated that insulin action on Akt phosphorylation and PI3K activity is unaltered in skeletal muscle of human subjects with newly diagnosed type 2 diabetes after treatment of rosiglitazone, another member of the TZD family. This discrepancy could be due to either the nature of the human subjects (obese type 2 diabetic vs. lean type 2 diabetic) or the different kinds of TZDs. From the view of the current human data, it is somewhat unlikely that changes in insulin signaling can fully account for the improvement of insulin sensitivity and glucose disposal in skeletal muscle in response to TZD treatment.
Given that correlative changes in fatty acid metabolism and improvements in glucose homeostasis and insulin sensitivity may imply an indirect effect on skeletal muscle via adipose tissue [63,64], it is possible that some of the effects of TZDs on insulin signaling work through secondary mechanisms. One potential factor is fatty acids, as elevations of fatty acids in plasma and the lipid content of muscle are associated with insulin resistance [65]. As previously reported [66,67], troglitazone treatment tends to lower triglyceride and free fatty acid concentrations. TZDs also reduce accumulation of muscle triglycerides and diacylglycerol [68]. Activation of PKC by elevated diacylglycerol levels in muscle impairs insulin signaling [69,70]. This raises the possibility that decreases in plasma lipid concentrations with TZD treatment could lead to a reduction of diacylglycerol in muscle, reducing PKC activation, resulting in an improvement of the insulin signaling cascade. It is also possible that TZDs reduce intramyocellular lipid content by promoting storage of free fatty acids in adipocyte triglycerides via PPARγ, redirecting free fatty acids from skeletal muscle to adipose tissue. Thus, the ability of troglitazone to improve insulin action in skeletal muscle could involve actions on both adipose tissue and muscle.
Metformin is a member of another class of drugs that are effective in lowering glucose concentrations in patients with type 2 diabetes [71]. A number of studies have demonstrated that metformin inhibits gluconeogenesis, reduces hepatic glucose output, and lowers fasting blood glucose concentration [59,72]. In addition to its effect on the liver, metformin also appears to decrease glucose concentrations by increasing peripheral insulin sensitivity and augmenting insulin-mediated glucose uptake in skeletal muscle of type 2 diabetic subjects [73]. The precise mechanism for this action of metformin is incompletely understood, but in vitro studies indicate it could involve multiple effects, including increased translocation of Glut1 and Glut4 glucose transporters from intracellular vesicles to the cell surface [74] and increased binding of insulin to cell surface insulin receptors [75]. Evidence shows that metformin normalizes insulin receptor tyrosine phosphorylation and PI3K and Akt activity in adipocytes exposed to high insulin levels in vitro for long periods [76]. However, our studies have demonstrated that the effects of metformin on PI3K and Akt activity in skeletal muscle of human insulin resistant subjects with obesity and type 2 diabetes in vivo are unchanged [60]. These data are further confirmed in skeletal muscle of patients with newly diagnosed type 2 diabetes [62]. Overall, it is clear that enhanced insulin-mediated glucose disposal by metformin therapy in type 2 diabetic patients is independent of improved insulin signaling in skeletal muscle of type 2 diabetic subjects.
Effects of weight loss therapy on skeletal muscle insulin signaling
Given that obesity is the major risk factor for developing insulin resistance, it is no doubt that lifestyle intervention that produces weight loss, improves insulin sensitivity. A reduction of fat mass by weight loss results in a significant decrease in lipid oxidation and an enhanced glucose homeostasis [77]. In addition, insulin secretion and plasma insulin concentrations decrease significantly after weight loss. Our work has investigated whether weight loss therapy can alter the ability of insulin to activate PI3K and downstream signaling in skeletal muscle of obese nondiabetic patients. For these studies, ten obese nondiabetic subjects (body mass index [BMI], 30 to 45 kg/m2) were challenged on a very low calorie diet (VLCD) of 600 to 800 calories per day for up to 24 weeks or until 10 to 15% of initial body weight was lost [78]. Once the weight loss goal was met, the subjects were introduced to a weight-maintenance diet. Glucose disposal rate measured by euglycemic hyperinsulinemic clamp was increased by 30% after VLCD treatment, indicating enhanced systemic insulin sensitivity. VLCD treatment significantly increased IRS-1 tyrosine phosphorylation, compared with the pretreatment level [78]. In parallel, insulin-stimulated IRS-1-associated PI3K activity was increased 2-fold post-treatment with VLCD, compared with pretreatment [78]. These changes were independent of the total amount of IRS-1 protein. Importantly, the impaired PKCλ/ζ activation in obese nondiabetic humans was reversed with weight reduction. This could be due to increased signaling upstream of PKCλ/ζ. This, combined with the observation that treating diabetic subjects with TZDs reverses the defect in PKCλ/ζ activity [79], suggests that reversal of the PKCλ/ζ defect might enhance insulin sensitivity in obese insulin-resistant humans. In line with these findings, recent studies of severely obese individuals also demonstrated that weight loss with a very low energy diet (1,883 kJ/day) markedly enhances insulin sensitivity by improving insulin-stimulated glucose disposal [77]. This enhanced sensitivity was accompanied by increased insulin signaling at the level of AS160 and proline-rich Akt substrate 40, which is a component of the mammalian target of the rapamycin nutrient-sensing pathway. Taken together, weight reduction in obese individuals improves insulin sensitivity, which may result from an improvement in PI3K and its downstream signaling in skeletal muscle.
Although our studies have not investigated Glut4 translocation in response to insulin in skeletal muscle of obese subjects that have undergone weight loss, a number of studies have demonstrated that enhancement in the insulin signaling pathway was not accompanied by a significant improvement in Glut4 translocation to the plasma membrane in skeletal muscle from type 2 diabetic patients, TZD-treated type 2 diabetic patients, and obese subjects who underwent gastric bypass surgery [62,80,81]. This could be explained by the fact that intrinsic activity of Glut4 at the plasma membrane is increased or that an unidentified glucose transporter, which may contribute to increased glucose disposal in skeletal muscle, is involved in this event.
Protein tyrosine phosphatase 1B and insulin signaling
Tyrosine phosphorylation of the IR is reversible, and IR dephosphorylation takes place rapidly in intact cells even with the continued presence of insulin [82]. Because a critical regulatory step in insulin signal transduction is the dephosphorylation of signaling molecules by PTPs, it is plausible that enhanced activity of one or more PTPs could lead to insulin resistance. Several studies of obese humans and rodents have reported that the expression and/or activity of specific PTPs, including the transmembrane LAR and the intracellular enzymes protein-tyrosine phosphatase 1B (PTP1B) and Src-homology 2 domain-containing phosphatase-2 (SHP2, SHPTP2, or syp), increase in muscle and adipose tissue [83-87]. LAR and PTP1B show the greatest increases (3-fold in muscle, 2-fold in fat) [83,88]. There is a strong correlation between BMI and total PTP activity toward the IR in both skeletal muscle and adipose tissue from lean and obese subjects [83,88]. Recent studies have also observed increased levels of LAR and PTP1B expression in liver and muscle from obese rodents [89].
Support for the role of PTPs in the regulation of insulin action comes from transgenic and gene knockout studies. The Kahn group generated PTP1B null mice by targeted disruption of the ATG-coding exon [90]. Elchebly et al. [91] targeted exons 5 and 6 (Ex5/6-/-) and also obtained PTP1B-null mice. Both lines of mice have increased insulin sensitivity, manifested by enhanced insulin-stimulated phosphorylation of IR and IRS-1 in muscle and liver. PTP1B-deficient mice have reduced body fat and are protected from diet-induced obesity due, at least in part, to increased basal metabolic rate and total energy expenditure [90]. In addition, insulin-stimulated whole-body glucose disposal is enhanced in PTP1B-deficient mice [90]. Surprisingly, this effect is tissue-specific; insulin-stimulated glucose uptake is elevated in skeletal muscle but not in adipose tissue. These data suggest that overexpression of PTP1B in insulin-target tissues in vivo could contribute to insulin resistance. Consistent with this hypothesis, Zabolotny et al. [28] demonstrated that selective overexpression of PTP1B in skeletal muscle impairs insulin-stimulated PI3K activity and causes mild insulin resistance in vivo. Moreover, recent studies have indicated that liver-specific deletion of PTP1B improves insulin resistance and attenuates diet-induced endoplasmic reticulum stress [92]. Similar to these findings, Haj et al. [93] showed that liver-specific re-expression of PTP1B in PTP1B deficient mice leads to marked attenuation of their enhanced insulin sensitivity. In obese diabetic insulin resistant ob/ob and db/db mice, PTP1B antisense oligonucleotide (ASO) treatment reduces PTP1B protein and mRNA level in liver and fat, and normalizes plasma glucose levels along with improved glucose tolerance and insulin sensitivity [94,95]. Taken together, the current data indicate that inhibition of PTP1B in peripheral tissues may be useful for treating metabolic-related disorders such as obesity and type 2 diabetes. In fact, the development of PTP1B inhibitors has received much attention by the pharmaceutical industry. However, it has been difficult to identify a selective, safe, and effective PTP1B inhibitor, although a novel PTP1B inhibitor (JTT-551) has been suggested as a potential therapeutic agent [96].
Rho-kinase and IRS-1 serine phosphorylation
Rho-kinase (ROCK) is a serine/threonine protein kinase identified as a GTP-Rho-binding protein [97]. There are two isoforms, ROCK1 (also known as ROKβ) [98,99] and ROCK2 (also known as ROKα) [98,100]. ROCK participates in the insulin signaling network by interacting with IRS-1 [101,102]. Our work has demonstrated that inhibiting ROCK decreases insulin-stimulated IRS-1-associated PI3K activity in adipocytes and myotubes. This effect is mainly due to decreased tyrosine phosphorylation of the YXXM motif in IRS-1, which can lead to reduced interaction of IRS-1 with the p85 subunit of PI3K. Indeed, insulin-stimulated IRS-1 binding to the p85 regulatory subunit of PI3K is impaired in adipocytes expressing dominant negative ROCK [103]. By mass spectrometry analysis, we identified the serine residues of IRS-1 at serine 632/635, serine 936, and serine 972, all of which are phosphorylated by ROCK. Interestingly, these sites are close to the YMXM motif domain in IRS-1, which is the binding site of the p85 regulatory subunit of PI3K. Evidence indicates that serine phosphorylation of IRS-1 is a key regulator of insulin signaling [104]. However, the effects of phosphorylation of individual IRS-1 serine residues on insulin signaling appear to be complex and may be context dependent; they are still being defined. Studies of the effects of IRS-1 serine 632/635 phosphorylation on insulin action have yielded conflicting results. Our studies have demonstrated that replacing IRS serines 632 and 635 with alanine causes a significant inhibition of insulin-stimulated IRS-1 tyrosine phosphorylation and PI3K activity [103], suggesting a positive role for IRS-1 serine 632/635 in insulin action.
Our recent studies have demonstrated that global deletion of ROCK1 in mice results in whole-body insulin resistance and impaired skeletal muscle insulin signaling. These effects are independent of changes in body adiposity. Insulin-stimulated IRS-1 serine 632/635 phosphorylation and PI3K activity associated with IRS-1 or phosphotyrosine are impaired in skeletal muscle of ROCK1 deficient mice [105]. Impaired insulin-induced IRS-1 serine 632/635 phosphorylation also accompanies decreased PI3K and ROCK activation in skeletal muscle of obese, insulin-resistant or diabetic mice and rats (unpublished data), suggesting that impaired IRS-1 serine 632/635 phosphorylation may be an important mechanism contributing to the pathogenesis of insulin resistance in obesity. In addition, insulin-stimulated ROCK activity is impaired in skeletal muscle of obese diabetic mice and insulin-resistant humans with obesity and type 2 diabetes (unpublished data). Importantly, insulin-stimulated ROCK1 activity was positively correlated with glucose disposal rate, suggesting that defective ROCK1 activation may contribute to the pathogenesis of human insulin resistance. Thus, our studies identify ROCK1 as a novel player regulating insulin-mediated glucose metabolism in vivo. Further studies of ROCK1 and ROCK2 functions in different metabolic tissues will be needed to precisely delineate ROCK isoform functions that regulate tissue and whole-body insulin sensitivity and glucose homeostasis. The emergence of ROCK1 as an important regulator of insulin action could lead to new treatment approaches for obesity and type 2 diabetes.
CONCLUSION
The earliest defect in the development of type 2 diabetes is insulin resistance characterized by decreased glucose transport and metabolism in skeletal muscle. Studies with the skeletal muscle of type 2 diabetic humans demonstrate impaired insulin activation of the IRS-1/PI3K/Akt signaling pathway, which is a critical step in the regulation of glucose transport in response to insulin. These defects are selectively restored by treatment with an insulin-sensitizing agent and lifestyle changes, representing the core of insulin signaling components. Recent advances have also revealed that insulin action on AS160 phosphorylation is diminished in skeletal muscle of type 2 diabetic patients, and that inhibiting AS160 causes a significant decrease in insulin-dependent translocation of Glut4, suggesting an important role for AS160 in glucose metabolism. Furthermore, defective ROCK activity in skeletal muscle may also contribute to impaired glucose homeostasis in type 2 diabetic patients. A better understanding of the disease pathogenesis and the potential alternative pathways for regulating glucose metabolism could lead to new therapeutic targets for obesity and type 2 diabetes by clarifying intracellular defects in the insulin signaling cascade.
No potential conflict of interest relevant to this article was reported.
1
DeFronzoRAPathogenesis of type 2 diabetes: metabolic and molecular implications for identifying diabetesDiabetes Rev19975177269
2
Yki-JärvinenHSahlinKRenJMKoivistoVALocalization of rate-limiting defect for glucose disposal in skeletal muscle of insulin-resistant type I diabetic patientsDiabetes1990391571672121571
3
PetersenKFShulmanGIPathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitusAm J Cardiol2002905A11G18G
4
KahnBBLilly lecture 1995. Glucose transport: pivotal step in insulin actionDiabetes199645164416548866574
5
GarveyWTHuecksteadtTPMatthaeiSOlefskyJMRole of glucose transporters in the cellular insulin resistance of type II non-insulin-dependent diabetes mellitusJ Clin Invest198881152815363366906
6
PedersenOBakJFAndersenPHEvidence against altered expression of GLUT1 or GLUT4 in skeletal muscle of patients with obesity or NIDDMDiabetes1990398658702354749
7
DohmGLEltonCWFriedmanJEDecreased expression of glucose transporter in muscle from insulin-resistant patientsAm J Physiol19912603 Pt 1E459E4632003599
8
KimYBNikoulinaSECiaraldiTPHenryRRKahnBBNormal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetesJ Clin Invest199910473374110491408
9
WhiteMFThe IRS-signaling system: a network of docking proteins that mediate insulin and cytokine actionRecent Prog Horm Res1998531191389769706
10
TaniguchiCMEmanuelliBKahnCRCritical nodes in signalling pathways: insights into insulin actionNat Rev Mol Cell Biol20067859616493415
11
SaleEMSaleGJProtein kinase B: signalling roles and therapeutic targetingCell Mol Life Sci20086511312717952368
12
FareseRVSajanMPStandaertMLInsulin-sensitive protein kinases (atypical protein kinase C and protein kinase B/Akt): actions and defects in obesity and type II diabetesExp Biol Med (Maywood)200523059360516179727
13
SanoHKaneSSanoEInsulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocationJ Biol Chem2003278145991460212637568
14
BandyopadhyayGSajanMPKanohYPKC-zeta mediates insulin effects on glucose transport in cultured preadipocyte-derived human adipocytesJ Clin Endocrinol Metab20028771672311836310
15
FareseRVSajanMPYangHMuscle-specific knockout of PKC-lambda impairs glucose transport and induces metabolic and diabetic syndromesJ Clin Invest20071172289230117641777
16
BourdeauADubéNTremblayMLCytoplasmic protein tyrosine phosphatases, regulation and function: the roles of PTP1B and TC-PTPCurr Opin Cell Biol20051720320915780598
17
HarleyEALevensNProtein tyrosine phosphatase 1B inhibitors for the treatment of type 2 diabetes and obesity: recent advancesCurr Opin Investig Drugs2003411791189
18
VinciguerraMFotiMPTEN and SHIP2 phosphoinositide phosphatases as negative regulators of insulin signallingArch Physiol Biochem20061128910416931451
19
SasaokaTWadaTTsunekiHLipid phosphatases as a possible therapeutic target in cases of type 2 diabetes and obesityPharmacol Ther200611279980916842857
20
CheathamBVlahosCJCheathamLPhosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocationMol Cell Biol199414490249118007986
21
Le Marchand-BrustelYGautierNCormontMVan ObberghenEWortmannin inhibits the action of insulin but not that of okadaic acid in skeletal muscle: comparison with fat cellsEndocrinology1995136356435707628394
22
BjörnholmMKawanoYLehtihetMZierathJRInsulin receptor substrate-1 phosphorylation and phosphatidylinositol 3-kinase activity in skeletal muscle from NIDDM subjects after in vivo insulin stimulationDiabetes1997465245279032113
23
KonishiHShinomuraTKurodaSOnoYKikkawaUMolecular cloning of rat RAC protein kinase alpha and beta and their association with protein kinase C zetaBiochem Biophys Res Commun19942058178257999118
24
KonishiHKurodaSTanakaMMolecular cloning and characterization of a new member of the RAC protein kinase family: association of the pleckstrin homology domain of three types of RAC protein kinase with protein kinase C subspecies and beta gamma subunits of G proteinsBiochem Biophys Res Commun19952165265347488143
25
KimYBPeroniODFrankeTFKahnBBDivergent regulation of Akt1 and Akt2 isoforms in insulin target tissues of obese Zucker ratsDiabetes20004984785610905496
26
WalkerKSDeakMPatersonAActivation of protein kinase B beta and gamma isoforms by insulin in vivo and by 3-phosphoinositide-dependent protein kinase-1 in vitro: comparison with protein kinase B alphaBiochem J19983312993089512493
27
JiangZYZhouQLColemanKAInsulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencingProc Natl Acad Sci U S A20031007569757412808134
28
ZabolotnyJMHajFGKimYBTransgenic overexpression of protein-tyrosine phosphatase 1B in muscle causes insulin resistance, but overexpression with leukocyte antigen-related phosphatase does not additively impair insulin actionJ Biol Chem2004279248442485115031294
29
ZabolotnyJMKimYBPeroniODOverexpression of the LAR (leukocyte antigen-related) protein-tyrosine phosphatase in muscle causes insulin resistanceProc Natl Acad Sci U S A2001985187519211309481
30
KimYBZhuJSZierathJRGlucosamine infusion in rats rapidly impairs insulin stimulation of phosphoinositide 3-kinase but does not alter activation of Akt/protein kinase B in skeletal muscleDiabetes19994831032010334307
31
KrookARothRAJiangXJZierathJRWallberg-HenrikssonHInsulin-stimulated Akt kinase activity is reduced in skeletal muscle from NIDDM subjectsDiabetes199847128112869703329
32
RondinoneCMCarvalhoEWesslauCSmithUPImpaired glucose transport and protein kinase B activation by insulin, but not okadaic acid, in adipocytes from subjects with Type II diabetes mellitusDiabetologia19994281982510440123
33
ChoHThorvaldsenJLChuQFengFBirnbaumMJAkt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in miceJ Biol Chem2001276383493835211533044
34
ChoHMuJKimJKInsulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta)Science20012921728173111387480
35
CeresaBPKaoAWSantelerSRPessinJEInhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathwaysMol Cell Biol199818386238709632770
36
KaneSSanoHLiuSCA method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domainJ Biol Chem2002277221152211811994271
37
NagaseTIshikawaKMiyajimaNPrediction of the coding sequences of unidentified human genes. IX. The complete sequences of 100 new cDNA clones from brain which can code for large proteins in vitroDNA Res1998531399628581
38
MorrisNJRossSALaneWSSortilin is the major 110-kDa protein in GLUT4 vesicles from adipocytesJ Biol Chem1998273358235879452485
39
SimpsonFWhiteheadJPJamesDEGLUT4--at the cross roads between membrane trafficking and signal transductionTraffic2001221111208163
40
BrussMDAriasEBLienhardGECarteeGDIncreased phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle in response to insulin or contractile activityDiabetes200554415015616009
41
DeshmukhACoffeyVGZhongZExercise-induced phosphorylation of the novel Akt substrates AS160 and filamin A in human skeletal muscleDiabetes2006551776178216731842
42
GeraghtyKMChenSHarthillJERegulation of multisite phosphorylation and 14-3-3 binding of AS160 in response to IGF-1, EGF, PMA and AICARBiochem J200740723124117617058
43
SriwijitkamolAColettaDKWajcbergEEffect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response studyDiabetes20075683684817327455
44
TreebakJTGlundSDeshmukhAAMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunitsDiabetes2006552051205816804075
45
HojlundKGlintborgDAndersenNRImpaired insulin-stimulated phosphorylation of Akt and AS160 in skeletal muscle of women with polycystic ovary syndrome is reversed by pioglitazone treatmentDiabetes20085735736617977950
46
KarlssonHKZierathJRKaneSInsulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjectsDiabetes2005541692169715919790
47
KotaniKOgawaWMatsumotoMRequirement of atypical protein kinase c lambda for insulin stimulation of glucose uptake but not for Akt activation in 3T3-L1 adipocytesMol Cell Biol199818697169829819385
48
BandyopadhyayGKanohYSajanMPStandaertMLFareseRVEffects of adenoviral gene transfer of wild-type, constitutively active, and kinase-defective protein kinase C-lambda on insulin-stimulated glucose transport in L6 myotubesEndocrinology20001414120412711089544
49
BandyopadhyayGStandaertMLGallowayLMoscatJFareseRVEvidence for involvement of protein kinase C (PKC)-zeta and noninvolvement of diacylglycerol-sensitive PKCs in insulin-stimulated glucose transport in L6 myotubesEndocrinology1997138472147319348199
50
BandyopadhyayDKusariAKennerKAProtein-tyrosine phosphatase 1B complexes with the insulin receptor in vivo and is tyrosine-phosphorylated in the presence of insulinJ Biol Chem1997272163916458999839
51
EtgenGJValasekKMBroderickCLMillerARIn vivo adenoviral delivery of recombinant human protein kinase C-zeta stimulates glucose transport activity in rat skeletal muscleJ Biol Chem1999274221392214210428775
52
BandyopadhyayGStandaertMLSajanMPDependence of insulin-stimulated glucose transporter 4 translocation on 3-phosphoinositide-dependent protein kinase-1 and its target threonine-410 in the activation loop of protein kinase C-zetaMol Endocrinol1999131766177210517677
53
KanohYBandyopadhyayGSajanMPStandaertMLFareseRVThiazolidinedione treatment enhances insulin effects on protein kinase C-zeta/lambda activation and glucose transport in adipocytes of nondiabetic and Goto-Kakizaki type II diabetic ratsJ Biol Chem2000275166901669610749857
54
KanohYBandyopadhyayGSajanMPStandaertMLFareseRVRosiglitazone, insulin treatment, and fasting correct defective activation of protein kinase C-zeta/lambda by insulin in vastus lateralis muscles and adipocytes of diabetic ratsEndocrinology20011421595160511250941
55
VollenweiderPMénardBNicodPInsulin resistance, defective insulin receptor substrate 2-associated phosphatidylinositol-3' kinase activation, and impaired atypical protein kinase C (zeta/lambda) activation in myotubes from obese patients with impaired glucose toleranceDiabetes2002511052105911916925
56
StandaertMLOrtmeyerHKSajanMPSkeletal muscle insulin resistance in obesity-associated type 2 diabetes in monkeys is linked to a defect in insulin activation of protein kinase C-zeta/lambda/iotaDiabetes2002512936294312351430
57
MudaliarSHenryRRNew oral therapies for type 2 diabetes mellitus: The glitazones or insulin sensitizersAnnu Rev Med20015223925711160777
58
KerstenSDesvergneBWahliWRoles of PPARs in health and diseaseNature200040542142410839530
59
InzucchiSEMaggsDGSpollettGREfficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitusN Engl J Med19983388678729516221
60
KimYBCiaraldiTPKongATroglitazone but not metformin restores insulin-stimulated phosphoinositide 3-kinase activity and increases p110beta protein levels in skeletal muscle of type 2 diabetic subjectsDiabetes20025144344811812753
61
StorgaardHSongXMJensenCBInsulin signal transduction in skeletal muscle from glucose-intolerant relatives of type 2 diabetic patients [corrected]Diabetes2001502770277811723060
62
KarlssonHKHällstenKBjörnholmMEffects of metformin and rosiglitazone treatment on insulin signaling and glucose uptake in patients with newly diagnosed type 2 diabetes: a randomized controlled studyDiabetes2005541459146715855334
63
ReginatoMJLazarMAMechanisms by which thiazolidinediones enhance insulin actionTrends Endocrinol Metab19991091310322388
64
KintscherULawREPPARgamma-mediated insulin sensitization: the importance of fat versus muscleAm J Physiol Endocrinol Metab2005288E287E29115637349
65
RodenMPriceTBPerseghinGMechanism of free fatty acid-induced insulin resistance in humansJ Clin Invest199697285928658675698
66
AntonucciTWhitcombRMcLainRLockwoodDNorrisRMImpaired glucose tolerance is normalized by treatment with the thiazolidinedione troglitazoneDiabetes Care1997201881939118772
67
PetersenKFKrssakMInzucchiSMechanism of troglitazone action in type 2 diabetesDiabetes20004982783110905493
68
Schmitz-PeifferCOakesNDBrowneCLKraegenEWBidenTJReversal of chronic alterations of skeletal muscle protein kinase C from fat-fed rats by BRL-49653Am J Physiol19972735 Pt 1E915E9219374677
69
LewisREVolleDJSandersonSDPhorbol ester stimulates phosphorylation on serine 1327 of the human insulin receptorJ Biol Chem199426926259262667929343
70
De FeaKRothRAProtein kinase C modulation of insulin receptor substrate-1 tyrosine phosphorylation requires serine 612Biochemistry19973612939129479335553
StumvollMNurjhanNPerrielloGDaileyGGerichJEMetabolic effects of metformin in non-insulin-dependent diabetes mellitusN Engl J Med19953335505547623903
73
DunnCJPetersDHMetformin. A review of its pharmacological properties and therapeutic use in non-insulin-dependent diabetes mellitusDrugs1995497217497601013
74
MatthaeiSHamannAKleinHHAssociation of Metformin's effect to increase insulin-stimulated glucose transport with potentiation of insulin-induced translocation of glucose transporters from intracellular pool to plasma membrane in rat adipocytesDiabetes1991408508571647995
75
FantusIGBrosseauRMechanism of action of metformin: insulin receptor and postreceptor effects in vitro and in vivoJ Clin Endocrinol Metab1986638989053745404
76
PryorPRLiuSCClarkAEChronic insulin effects on insulin signalling and GLUT4 endocytosis are reversed by metforminBiochem J2000348Pt 1839110794717
77
JazetIMSchaartGGastaldelliALoss of 50% of excess weight using a very low energy diet improves insulin-stimulated glucose disposal and skeletal muscle insulin signalling in obese insulin-treated type 2 diabetic patientsDiabetologia20085130931918080107
78
KimYBKotaniKCiaraldiTPHenryRRKahnBBInsulin-stimulated protein kinase C lambda/zeta activity is reduced in skeletal muscle of humans with obesity and type 2 diabetes: reversal with weight reductionDiabetes2003521935194212882908
79
BeesonMSajanMPDizonMActivation of protein kinase C-zeta by insulin and phosphatidylinositol-3,4,5-(PO4)3 is defective in muscle in type 2 diabetes and impaired glucose tolerance: amelioration by rosiglitazone and exerciseDiabetes2003521926193412882907
80
RyderJWYangJGaluskaDUse of a novel impermeable biotinylated photolabeling reagent to assess insulin- and hypoxia-stimulated cell surface GLUT4 content in skeletal muscle from type 2 diabetic patientsDiabetes20004964765410871204
81
FriedmanJEDohmGLLeggett-FrazierNRestoration of insulin responsiveness in skeletal muscle of morbidly obese patients after weight loss. Effect on muscle glucose transport and glucose transporter GLUT4J Clin Invest1992897017051737857
82
GoldsteinBJAhmadFDingWLiPMZhangWRRegulation of the insulin signalling pathway by cellular protein-tyrosine phosphatasesMol Cell Biochem199818291999609118
83
AhmadFConsidineRVGoldsteinBJIncreased abundance of the receptor-type protein-tyrosine phosphatase LAR accounts for the elevated insulin receptor dephosphorylating activity in adipose tissue of obese human subjectsJ Clin Invest199595280628127769120
84
AhmadFGoldsteinBJIncreased abundance of specific skeletal muscle protein-tyrosine phosphatases in a genetic model of insulin-resistant obesity and diabetes mellitusMetabolism199544117511847666792
85
AhmadFGoldsteinBJFunctional association between the insulin receptor and the transmembrane protein-tyrosine phosphatase LAR in intact cellsJ Biol Chem19972724484578995282
86
AhmadFGoldsteinBJEffect of tumor necrosis factor-alpha on the phosphorylation of tyrosine kinase receptors is associated with dynamic alterations in specific proteintyrosine phosphatasesJ Cell Biochem1997641171279015760
87
McGuireMCFieldsRMNyombaBLAbnormal regulation of protein tyrosine phosphatase activities in skeletal muscle of insulin-resistant humansDiabetes1991409399421647997
88
AhmadFAzevedoJLCortrightRDohmGLGoldsteinBJAlterations in skeletal muscle protein-tyrosine phosphatase activity and expression in insulin-resistant human obesity and diabetesJ Clin Invest19971004494589218523
89
ZabolotnyJMKimYBWelshLAProtein-tyrosine phosphatase 1B expression is induced by inflammation in vivoJ Biol Chem2008283142301424118281274
90
KlamanLDBossOPeroniODIncreased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient miceMol Cell Biol2000205479548910891488
91
ElcheblyMPayettePMichaliszynEIncreased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B geneScience19992831544154810066179
92
DelibegovicMZimmerDKauffmanCLiver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stressDiabetes20095859059919074988
RondinoneCMTrevillyanJMClampitJProtein tyrosine phosphatase 1B reduction regulates adiposity and expression of genes involved in lipogenesisDiabetes2002512405241112145151
95
ZinkerBARondinoneCMTrevillyanJMPTP1B antisense oligonucleotide lowers PTP1B protein, normalizes blood glucose, and improves insulin sensitivity in diabetic miceProc Natl Acad Sci U S A200299113571136212169659
96
FukudaSOhtaTSakataSPharmacological profiles of a novel protein tyrosine phosphatase 1B inhibitor, JTT-551Diabetes Obes Metab20101229930620380650
97
MatsuiTAmanoMYamamotoTRho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein RhoEMBO J199615220822168641286
98
NakagawaOFujisawaKIshizakiTROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in miceFEBS Lett19963921891938772201
99
IshizakiTMaekawaMFujisawaKLimLThe small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinaseEMBO J199615188518938617235
100
LeungTManserETanLA novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranesJ Biol Chem199527029051290547493923
101
FarahSAgazieYOhanNNgseeJKLiuXJA rho-associated protein kinase, ROKalpha, binds insulin receptor substrate-1 and modulates insulin signalingJ Biol Chem1998273474047469468537
102
BegumNSanduOAItoMLohmannSMSmolenskiAActive Rho kinase (ROK-alpha) associates with insulin receptor substrate-1 and inhibits insulin signaling in vascular smooth muscle cellsJ Biol Chem20022776214622211739394
103
FurukawaNOngusahaPJahngWJRole of Rho-kinase in regulation of insulin action and glucose homeostasisCell Metab2005211912916098829
104
ZickYInsulin resistance: a phosphorylation-based uncoupling of insulin signalingTrends Cell Biol20011143744111684411
105
LeeDHShiJJeoungNHTargeted disruption of ROCK1 causes insulin resistance in vivoJ Biol Chem2009284117761178019276091
Figure 1
The insulin signaling pathway. PTP1B, protein-tyrosine phosphatase 1B; IRS, insulin receptor substrate; ROCK, Rho-kinase; PIP, phosphatidylinositol phosphate; PTEN, phosphatase and tension homologue deleted on chromosome 10; PH domain, pleckstrin homology domain; PDK, phosphoinositide-dependent protein kinase; GβL, G-protein beta subunit like; mTOR, mammalian target of rapamycin; AS160, 160 kDa Akt substrate; PKCλ/ζ, protein kinase C λ and ζ; Glut4, glucose transporter 4.