24-Hour blood pressure response to lower dose (30 mg) fimasartan in Korean patients with mild to moderate essential hypertension
Article information
Abstract
Background/Aims
Fimasartan is an angiotensin type 1 receptor blocker (ARB) which has comparable efficacy and tolerability with other ARBs. The aim of this study was to evaluate 24-hour blood pressure (BP) lowering efficacy and the tolerability of the low dose fimasartan compared with valsartan in patients with mild to moderate hypertension.
Methods
This study was a phase II, prospective, multicenter, randomized, double-blind, parallel-grouped trial. A total of 75 hypertensive patients, whose mean ambulatory BP monitoring values were ≥ 135/85 mmHg, were randomized to either fimasartan 30 mg or valsartan 80 mg daily. The primary efficacy endpoint was the change in the mean 24-hour systolic BP (SBP) values from the baseline and at the week 8. Secondary endpoints included the change in the mean 24-hour diastolic BP values, the daytime and the nighttime mean BP values at week 8, the trough-to-peak (T/P) ratio and the smoothness index.
Results
At week 8, the mean 24-hour SBP values significantly decreased in both groups; –10.5 ± 11.9 mmHg (p < 0.0001) in the fimasartan group and –5.5 ± 11.6 mmHg (p = 0.0307) in the valsartan group. The difference between two groups was 4.3 ± 2.9 mmHg but there was no statistical significance (p = 0.1392). The global T/P ratio in the fimasartan 30 mg groups were 0.48 and 0.40 in the valsartan 80 mg group, respectively (p = 0.3411). The most frequent adverse events (AEs) were acute pharyngitis and there were no cases of severe AEs.
Conclusions
In mild-to-moderate hypertensive patients, low dose (30 mg) fimasartan showed comparable 24-hour BP lowering efficacy compared with valsartan (80 mg). There was no difference in tolerability between two groups.
INTRODUCTION
Hypertension is the most common cause of cardiovascular disease [1]. Blood pressure (BP) control by antihypertensive medication is effective strategy to reduce cardiovascular morbidity and mortality [2,3]. Among the different classes of antihypertensive drugs, angiotensin type 1 receptor blockers (ARBs), which regulate the renin-angiotensin-aldosterone system by restricting the action of angiotensin II, are recommended as a first-line therapy in all guidelines [4-8].
Fimasartan, developed by Boryung Pharmaceutical Co. Ltd., Seoul, Korea, is the eighth ARB out of nine currently available ARBs [9,10]. Fimasartan at the range of 30 to 120 mg once daily showed an effective BP lowering effect in hypertensive patients [11-14]. Also in large population observation study, fimasartan showed excellent safety profile [15,16]. Several pharmacokinetic/pharmacodynamic studies were performed to evaluate the possible drug or food interactions [17-20] and the drug metabolism in the elderly population and in hepatic dysfunction [21,22]. In patients with moderate hepatic impairments, the bioavailability of fimasartan was 5-fold higher than that of healthy controls [20,21]. The co-administration of fimasartan with amlodipine, hydrochlorothiazide, or digoxin did not show any significant interactions [23-25]. Conversely, fimasartan raised plasma atorvastatin concentrations [20].
Previously, fimasartan 60 mg was the minimum permitted dose for hypertension treatment. However, in real world situation, half tablet (30 mg) of fimasartan was often used and was suggested to have sufficient BP lowering efficacy. Also, fimasartan 30 mg showed superior office BP lowering efficacy than valsartan 80 mg in patients with mild to moderate hypertension [26]. With this background, this study was conducted to evaluate whether once daily administration of the low dose (30 mg) fimasartan is able to maintain BP lowering toward the end of the dosing interval. Ambulatory blood pressure monitor (ABPM) was performed to evaluate 24-hour BP lowering efficacy of the low dose fimasartan (30 mg once daily) compared with valsartan (80 mg once daily) in patients with mild to moderate hypertension.
METHODS
Patients
Male or non-childbearing female patients aged 20 to 70 with mild to moderate hypertension whose mean 24-hour ABPM values ≥ 135/85 mmHg were eligible for the study. For office BP measurement, patients should be seated at least for 5 minutes and refrain from smoking or ingesting caffeine during the 30 minutes prior to the measurement. Patients were excluded from the study if they met any of the following exclusion criteria: (1) severe hypertension defined as mean sitting diastolic blood pressure (SiDBP) ≥ 110 mmHg or mean sitting systolic blood pressure (SiSBP) ≥ 180 mmHg at the screening, baseline and randomization visits; (2) a change in SiDBP ≥ 10 mmHg or a change in SiSBP ≥ 20 mmHg measured in the same arm at the screening visit; (3) secondary hypertension including pheochromocytoma, renal artery stenosis, and functional adrenal adenoma; (4) symptomatic orthostatic hypotension; (5) uncontrolled diabetes before the screening visit (glycosylated hemoglobin > 9%), current treatment with insulin, or a change in oral hypoglycemic agent dosage/usage within the 12 weeks before the screening visit; (6) history of myocardial infarction or coronary arterial disease, clinically significant congestive heart failure or valvular heart disease identified within the past six months; and (7) serum creatinine ≥ 1.5 times upper normal limit (< 1.40 mg/dL), aspartate aminotransferase or alanine aminotransferase ≥ 2 times upper normal limit (< 40 IU/L), and other clinically significant abnormal laboratory test results.
Study design
This study was a phase II, prospective, multicenter, randomized, double-blind, parallel-grouped trial to compare 24-hour BP lowering efficacy and the tolerability of the low-dose fimasartan and valsartan in patients with mild to moderate essential hypertension. All of the study protocols were reviewed and approved by the Institutional Review Board of each participating hospital prior to any patient enrollment (IRB No. 1102-072-352). In addition, the study was conducted in accordance with the current Good Clinical Practices and other applicable laws and regulatory requirements in Korea. This study was registered in Clinicaltrials.gov (NCT01878201).
Subjects went through a screening period after voluntary agreement to participate in the study including a 7-day washout period for those patients who had been taking other medication. After 2 weeks’ placebo run-in, the eligible hypertensive patients, whose mean ABPM values were ≥ 135/85 mmHg, were randomized to either fimasartan 30 mg daily or valsartan 80 mg daily. Patients were instructed to orally take the assigned drug once daily at the same time after a meal in the morning while they came to the clinic without taking the drug when study procedures were scheduled at weeks 0, 4, and 8. During these study visits, BP measurements, physical examinations, clinical laboratory tests (i.e., hematology, blood chemistry, and urinalysis), and 12-lead electrocardiogram (ECG) were performed.
Compliance was assessed by investigators based on the rate of return and amount of administration of investigational products. Clinical pharmacists recorded the amount of returned product when a subject delivered the remaining product to pharmacist. Compliance was assessed as below: compliance (%) = (total number of administered tablets / total number of tablets to be administered according to the protocol) × 100. The study design was summarized in Fig. 1.
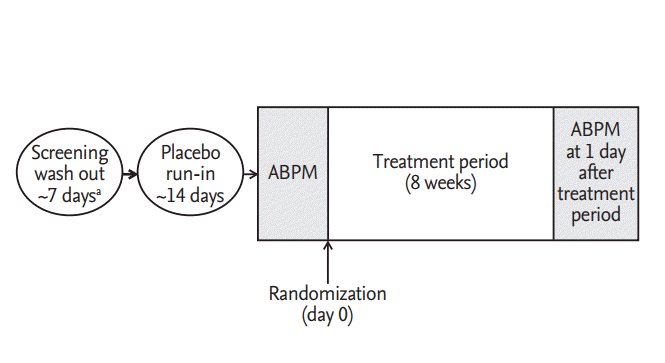
Study design. ABPM, ambulatory blood pressure monitor. a Previous anti-hypertensive medication users only.
24-Hour ambulatory blood pressure monitoring
We used the same protocol as previous ABPM study with fimasartan 20 to 180 mg [11,12]. Every center used same ABPM device of HEM-7080IT (equivalent to 705IT, Omron, Kyoto, Japan). With patients reporting at the clinic in the morning, ABPM started and continued for the subsequent 24 hours. During ABPM, patients were instructed to record daily events and were discouraged from engaging in strenuous physical activities. Patients were also informed in advance of the possible problems related to ABPM and instructed how to stop ABPM when they were occurred. Ambulatory BP was recorded every 20 minutes from 7:00 AM to 10:59 PM (daytime) and every 30 minutes from 11:00 PM to 6:59 AM the next day (nighttime). BP recordings were accepted only if they spanned the full 24 hours and at least 80% of the expected recordings were recorded. Readings of SBP < 70 or > 250 mmHg and DBP < 40 or > 150 mmHg were discarded and daytime BPs were averaged over a 1-hour period. ABPs were then synchronized (i.e., matched by measurement time) in each patient and 8-week values were subtracted from baseline values to determine 24-hour, daytime, and nighttime mean changes.
The trough-to-peak (T/P) ratio was calculated to evaluate the sustainability of BP reduction toward the end of the dosing interval. The trough was defined as the ambulatory BP reduction over the last 2 hours of the dosing interval (i.e., 23 to 24 hours post-dose) [27]. The peak effect was defined as the maximal decrease in ambulatory BP from baseline over any 1-hour period after drug administration using 2-hour moving averages [27]. ABPM data were then matched by measurement time in each patient, and values at 8 weeks of treatment were subtracted from the baseline values to determine the trough and peak effects. From these effects, global T/P ratio was calculated from all patients for a single effect profile (global T/P ratio), as previously recommended [28,29].
The smoothness index (SI) was also calculated to evaluate all treatment effects over the full 24-hour period [30]. SI was calculated as the ratio of the mean of hourly BP changes to their standard deviation using the measurement time-matched ABPM data. A greater SI denotes lesser variability of BP reduction by treatment over the full dosing interval, although there is no reference value for SI [31].
End points
The primary efficacy endpoint was the change in the mean 24-hour SBP values from the baseline and at the week 8. Secondary endpoints included the change in the 24-hour mean DBP values from the baseline at the week 8, the daytime and the nighttime mean BP values at week 8, the T/P ratio and the SI of each drugs. T/P ratio analysis was performed individually and globally as previously recommended in the latter case of which all patients were combined to come up with a single effect profile, from which global estimates were determined [12,28].
Safety endpoints were treatment-emergent adverse events (TEAEs), which were defined as any untoward events not present prior to the administration of the study drug or already present but worsened in either intensity or frequency during treatment. TEAEs included abnormalities found in clinical laboratory tests, physical examinations, and ECG readings.
Sample size and statistical analysis
Since this study was initiated as an exploratory study in order to respond the practical need from the clinical field, we enrolled 35 patients in each group which was the minimum regulatory requirements in Korean governmental regulation for addition of the new dosage form. With an anticipated dropout rate of 10%, a total of 68 patients were required to fulfill the enrollment of 34 patients in two groups.
Efficacy data was evaluated mainly in full analysis set (FAS) group and also in the per-protocol (PP) group. The FAS population consisted of the subjects who had the baseline BP and ≥ one post-randomization BP values and who had taken ≥ one dose of study drug. The PP population consisted of all subjects within the FAS population who did not commit any major protocol violation and drop-out that would likely to affect the efficacy outcomes. The safety set population consisted of the group of subjects who received investigational products at least once or more after randomization.
Changes from the baseline in the mean 24-hour SBP/DBP, the daytime and the nighttime SBP/DBP at 8 weeks were analyzed by paired t test in each treatment group. The between-group differences were analyzed by analysis of covariance model using site and baseline as covariates [13]. Nonparametric tests such as the Wilcoxon rank sum test were also used when deemed necessary. The proportion of patients experiencing adverse events (AEs) and its 95% confidence interval were also presented by the treatment group and a chi-square test or Fisher exact test was performed for the between-group difference. SAS version 9.3 (SAS Institute, Inc., Cary, NC, USA) was used for statistical analysis.
RESULTS
Patient recruitment and baseline characteristics
In six institutions, 75 patients among 138 patients, who were screened and fulfilled the selection criteria, were randomly allocated into two treatment groups. During placebo run-in period, two patients withdrew consent and four patients were lost to follow-up. Among 75 patients who were randomized and took a single dose of the study drug, 67 patients completed the study. The reasons for discontinuation were as followed; withdrawal of consent (three patients), protocol deviations (three patients), and investigators’ decision of safety concern for BP elevation (two patients). Patient recruitment and flow were summarized in Fig. 2.
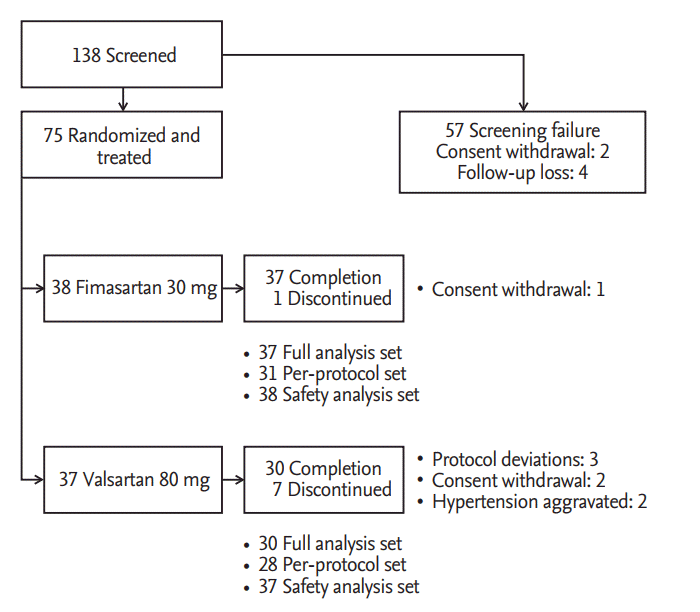
Patients recruitment and flow.
Two treatment groups were comparable for all baseline demographic and clinical characteristics except higher BMI in the fimasartan group (Table 1). The mean ± SE age was 57.1 ± 7.6 years old and the proportion of male patients was 70.2%. Mean baseline office SBP/DBP were 147.4 ± 12.6/90.4 ± 8.6 mmHg. The 73.1% of patients had been previously treated with anti-hypertensive agents, most frequently with ARBs (37.3%), calcium channel blockers (32.8%), and β-blockers (7.5%). There was no significant difference in previous anti-hypertensive medication history between two groups (Table 1).
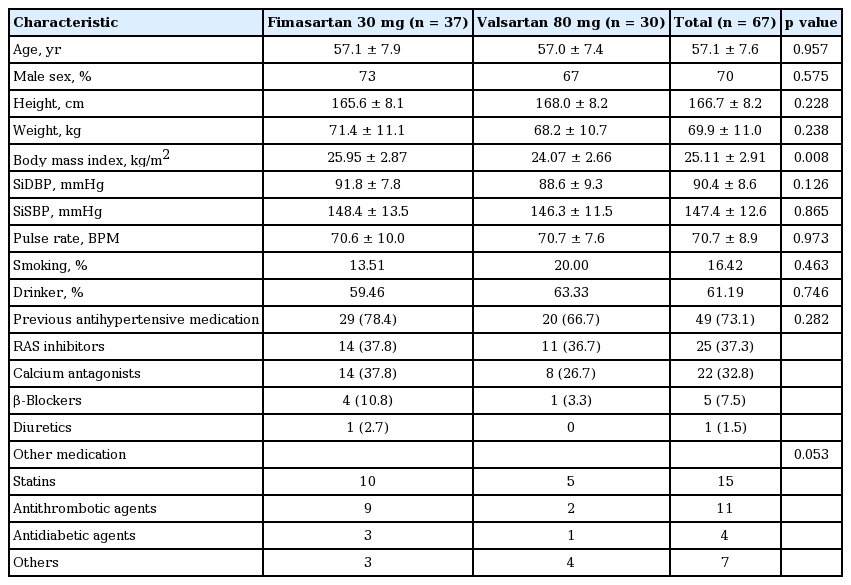
Baseline demographic and clinical characteristics
Efficacy outcomes
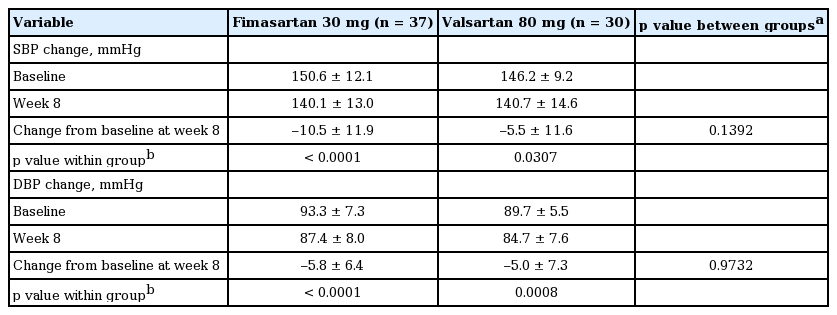
At week 8, the mean 24-hour SBP significantly decreased from the baseline in both groups; –10.5 ± 11.9 mmHg in the fimasartan group (p < 0.0001) (Fig. 3A) and –5.5 ± 11.6 mmHg in the valsartan group (p = 0.0307) (Fig. 3B). The difference between two groups (least square mean ± SE) was 4.3 ± 2.9 mmHg but there was no statistical significance (p = 0.1392) (Fig. 3C). PP group showed similar results; –11.1 ± 12.6 mmHg in the fimasartan group (p < 0.0001) and –6.1 ± 11.7 mmHg in the valsartan group (p = 0.0108), but there was no statistical significant difference between two groups, either (p = 0.1552) (Table 2). The mean 24-hour DBP also significantly decreased from the baseline in both groups; –5.8 ± 6.4 mmHg (p < 0.0001) in the fimasartan group and –5.0 ± 7.3 mmHg (p = 0.0008) in the valsartan group, but there was no statistical significant difference between two groups, either (p = 0.9732).

24-Hour systolic blood pressure (SBP) profiles and change from the baseline at week 8. 24-Hour SBP profiles of (A) fimasartan 30 mg and (B) valsartan 30 mg by time after dosing. (C) Change from the baseline at week 8 in 24-hour SBP. Data were analyzed on full analysis set. CI, confidence C interval.
Change in the mean 24-hour blood pressures from the baseline at week 8 (full analysis set)
After 8 weeks’ treatment, both fimasartan 30 mg and valsartan 80 mg significantly reduced the daytime, and the nighttime mean SBP by –8.0 ± 12.1 mmHg and –4.1 ± 12.1 mmHg and –15.7 ± 14.9 mmHg and –8.0 ± 15.3 mmHg, respectively (Table 3).
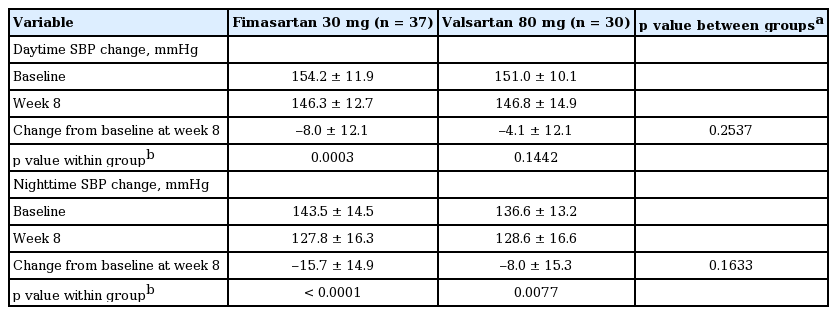
Change in daytime and nighttime SBPs from the baseline at week 8 (full analysis set)
The changes of the mean 24-hour, the daytime and the nighttime BPs from the baseline at week 8 were illustrated in Fig. 4.
The changes of the mean (A) 24-hour, (B) the daytime, and (C) the nighttime blood pressures (BPs) from the baseline at week 8. SBP, systolic blood pressure; DBP, diastolic blood pressure.
Fimasartan 30 mg daily was associated with the similar BP reduction toward the end of the dosing interval compared with valsartan 80 mg daily, which resulted in statistically insignificant difference of T/P ratio. The global T/P ratios for ambulatory SBP and DBP were 0.48 and 0.34 in the fimasartan group and 0.40 and 0.52 in the valsartan group (p = 0.3411 for SBP, p = 0.7288 for DBP). The SIs for ambulatory SBP and DBP were 0.86 and 0.63 in the fimasartan group and 0.51 and 0.58 in the valsartan group (p = 2538 for SBP, p = 0.7885 for DBP) (Table 4).
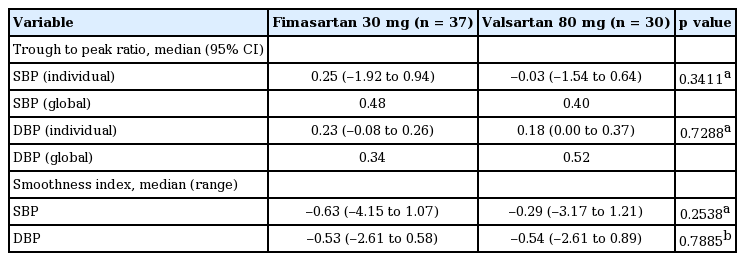
Trough to peak ratio and smoothness index in SBP and DBP (full analysis set)
Tolerability
Both group showed excellent compliance rate of 96.6% ± 4.0% (96.6 ± 3.7 vs. 96.2 ± 4.3, p = 0.8418). Although we collected all the activity and the event during ABPM measurement, there were no remarkable events reported. The proportions of patients experiencing ≥ 1 TEAE were comparable between two groups; four patients (five events, 10.5%) in the fimasartan group and six patients (eight events, 16.2%) in the valsartan group (p = 0.5161). No serious TEAEs were reported, and acute pharyngitis (one patient in the fimasartan group, two patients in the valsartan group) was the most commonly reported TEAE. In severity, 10 AEs were mild and three AEs were moderate; influenza (one case, fimasartan), tonsillitis (one case, valsartan), dizziness (one case, valsartan). Drug-related AEs did not occur in either group. And there was no case of serious AEs. There was no specific abnormality in laboratory test, vital sign, and physical examination (Table 5).
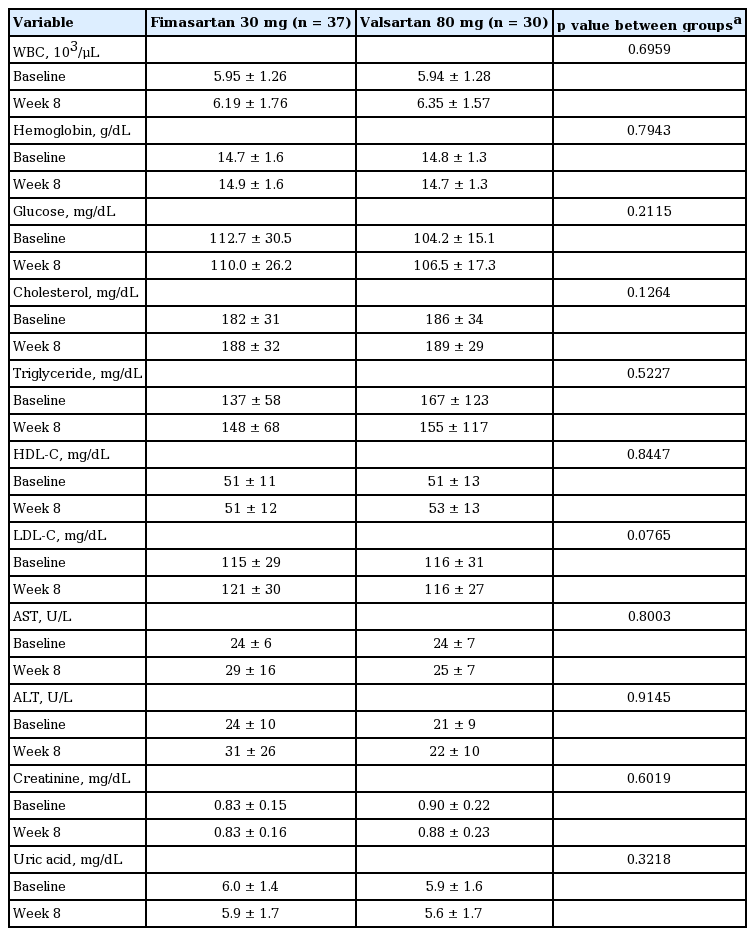
Key laboratory data change from the baseline at week 8 (full analysis set)
DISCUSSION
The objective of the present study was to compare 24-hour BP lowering efficacy of low dose (30 mg) fimasartan with valsartan (80 mg) in patients with mild to moderate hypertension. The results of this study indicate that the efficacy of low dose (30 mg) fimasartan showed comparable 24-hour BP lowering efficacy with valsartan (80 mg).
In the initial dose finding stage of drug development, fimasartan 20 mg did not show significantly different office BP lowering efficacy compared with the placebo. And a clear demarcation between fimasartan 20 and 60 mg was noted [12]. Therefore, fimasartan 60 mg was chosen as a minimum tablet size. However, in real world situation, half tablet, 30 mg of fimasartan was frequently used as an initial dose or during down-titration step. Therefore, in order to confirm that finding and to prepare the new dosage form, the efficacy study and ABPM study were conducted to evaluate whether low dose (30 mg) fimasartan once daily can reduce BP toward the end of the dosing interval and to compare its efficacy with valsartan 80 mg. In previous efficacy comparison study of fimasartan 30 mg with valsartan 80 mg, after 8 weeks treatment, fimasartan 30 mg showed superior office BP lowering efficacy than valsartan 80 mg in patients with mild to moderate hypertension [26].
ARBs are known to have flat dose-response relationship in BP lowering efficacy [32-34]. Previously, fimasartan 60mg daily was reported to have greater T/P ratios and SI values than valsartan 80 mg daily. Longer half-life of fimasartan, 10- to 17.9-hour [19], might explain greater sustainability of 24-hour BP reduction than valsartan which has an intermediate to long half-life of 6 to 10 hours [35,36]. However, increasing the ARB dose was reported to be effective in increasing the duration of the anti-hypertensive effect [37]. Conversely, lower dose of ARBs might have insufficient 24-hour lowering efficacy. Therefore, we could not conclude that lower dose fimasartan had sufficient 24-hour BP lowering efficacy. In this aspect, this study was performed to confirm whether low dose (30 mg) fimasartan maintained 24-hour BP lowering [11]. As expected, T/P ratios and SI values of lower dose fimasartan were both lower than those values reported in fimasartan 60 mg [11]. However, the values were still numerically greater than those of valsartan 80 mg.
Although this study did not measure the exact placebo-subtracted T/P ratio, T/P ratio of fimasartan 30 mg approximated but smaller than 0.5, which is the lower limit for approval of antihypertensive drugs administered once daily [38], even though recent guideline of the U.S. Food and Drug Administration becomes less strict and flexible [39]. Therefore we should note the possibility of BP fluctuation when prescribing lower dose of fimasartan. And if patients appeal the BP rise in evening or early in the morning, the up-titration of fimasartan dose to increase duration of action will be highly recommended.
The major limitation of this study was the small number of patients and short term duration of study. Therefore we could not confirm any support of superiority of fimasartan 30 mg daily to valsartan 80 mg with statistical significance even though the values were numerically greater than those of valsartan 80 mg.
In conclusion, in adult Korean patients who had mild-to-moderate hypertension, the efficacy of low dose (30 mg) fimasartan showed comparable 24-hour BP lowering efficacy compared with valsartan (80 mg). Fimasartan 30 mg effectively maintained BP reduction profile over the full 24-hour dosing interval, suitable for once daily dosing. Fimasartan was well tolerated.
KEY MESSAGE
1. Angiotensin type 1 receptor blockers are known to have flat dose-response relationship in blood pressure (BP) lowering efficacy.
2. The efficacy of low dose (30 mg) fimasartan showed comparable 24-hour BP lowering efficacy with valsartan (80 mg).
3. Fimasartan 30 mg effectively maintained BP reduction profile over the full 24-hour dosing interval, suitable for once daily dosing.
Notes
This study was sponsored by Boryung pharmaceutical, Seoul, Korea. The sponsor supported the supply of the investigational products, laboratory test, and clinical research coordinator expenses.
Acknowledgements
Drs. Byung-Hee Oh and Hae-Young Lee made substantial contributions to the study design. Drs. Byung-Hee Oh and Hae-Young Lee were contributed in the manuscript writing and figure creation. All the authors were equally contributed in the data collection, data interpretation, literature research, and involved in all stages of manuscript development.