Clinical significance of Th17 cells in kidney transplantation
Article information
Abstract
Transplantation research has focused on cytotoxic T-cell and plasma cell/B-cell-targeted strategies, but little attention has been paid to the role of T helper 17 (Th17) cells in allograft dysfunction. However, accumulating evidence suggests that Th17 cells contribute to the development of acute and chronic allograft injury after transplantation of various organs, including the kidney. This review summarizes recent reports on the role of Th17 cells in kidney transplantation. Means of improving allograft outcomes by targeting the Th17 pathway are also suggested.
INTRODUCTION
Kidney transplantation (KT) is the best available treatment option for end-stage renal disease. However, the long-term success rate of KT is limited by acute or chronic allograft rejection due to progressive destruction caused by recognition of donor alloantigens by the recipient’s immune system [1]. Allograft rejection after KT is mediated mainly by helper T-cells. The Th1–Th2 balance is regarded as the major mechanism of rejection [2]. However, certain immunologic events occurring after KT cannot be explained solely by the Th1–Th2 balance. Therefore, the mechanism of rejection has been expanded to include T helper 17 (Th17) cells, which secrete the proinflammatory cytokine interleukin 17 (IL-17) [3].
Th17 cells were identified as a subset distinct from T helper type 1 and 2 cells in 2005 [3-5]. Th17 cells were first found to be clinically important in autoimmune disorders, and are now of interest in transplantation [6-8]. Indeed, Th17 cells and their associated cytokines play an important role in the development of acute and chronic allograft injury after organ transplantation [1,8-12]. For example, IL-17 messenger RNA (mRNA) and protein levels are elevated in animal models of acute rejection [13]. In KT, the IL-17 mRNA level is elevated in proximal tubular epithelial cells from allograft tissue with subclinical rejection [14]. Moreover, the IL‑17 protein level is elevated, and IL-17 mRNA is detectable, in kidney allografts with subclinical rejection [7]. Here, we review the findings on the role of Th17 cells in the development of acute or chronic rejection in KT, and discuss strategies to control the Th17 pathway.
OVERVIEW OF TH17 CELLS
Th17 cells secrete IL-17, which recruits monocytes and neutrophils and acts in synergy with other proinflammatory cytokines [3]. Transforming growth factor β, IL-6, and IL-1β mediate the induction of immature Th17 cells [15,16]. However, they have a different nuclear transcript factor profile compared to classical Th1 cells; i.e., RORγt (RAR-related orphan receptor γt) and signal transducer and activator of transcription 3 (STAT3) [3,17]. Finally, differentiation into effector Th17 cells is mediated by the IL-23–IL-23R interaction [18]. Mature Th17 cells produce IL-22, express C-C motif chemokine receptor (CCR)-6, -4, and -10, and do not produce IL-10 (Fig. 1) [19-21].
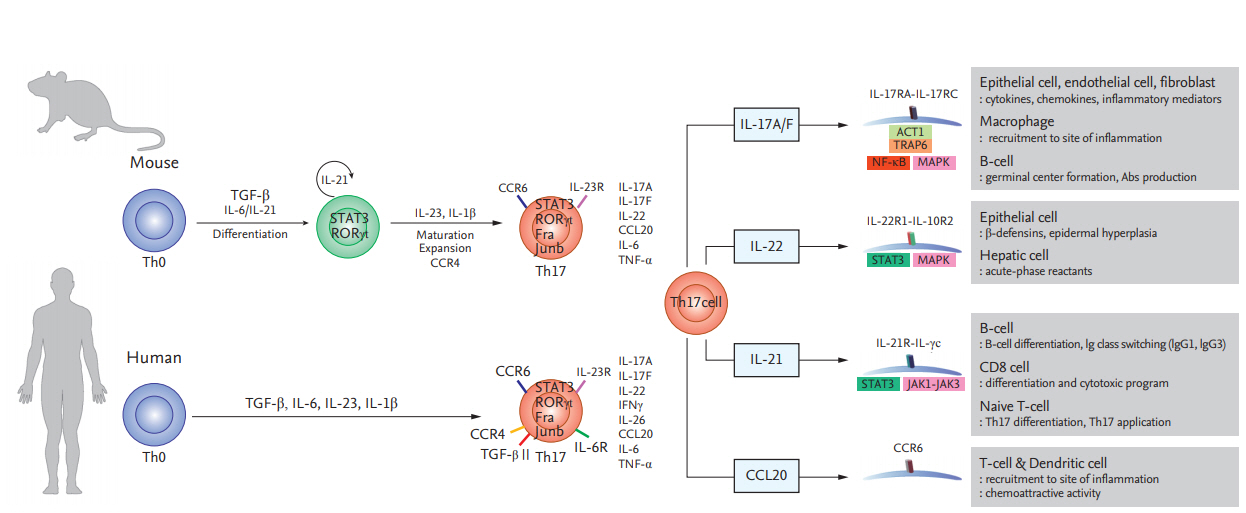
Differentiation of T helper 17 (Th17) cells in mice and humans and the functions of Th17 cytokines and chemokines. Th17 cells secrete interleukin 17A/F (IL-17A/F), interleukin 22 (IL-22), interleukin 21 (IL-21), and C-C motif chemokine ligand 20 (CCL20), which modulate inflammation and immune cell recruitment. TGF-β, Transforming growth factor β; STAT3, signal transducer and activator of transcription 3; RORγt, RAR-related orphan receptor γt; CCR, C-C motif chemokine receptor; ACT1, actin 1; TRAP6, thrombin receptor-activating peptide-6; NF-κB, nuclear factor κB; MAPK, mitogen-activated protein kinase; IgG, immunoglobulin G.
TH17 CELLS IN KIDNEY ALLOGRAFT TISSUE WITH ACUTE ALLOGRAFT REJECTION
Acute allograft rejection is initiated by alloreactive T-cells primed in secondary lymphoid organs and recruited to the graft. The production of various proinflammatory cytokines by infiltrating cells is increased during acute kidney allograft rejection [22-24]. Therefore, diagnosis and staging of allograft rejection are based on the severity or pattern of immune cell infiltration and evidence of local immune system activation in allograft tissue [25-28]. Increased expression of IL-17 in local tissue is associated with allograft rejection in vivo [7,14,29]. Also, greater infiltration of Th17 cells compared to FOXP3+ (forkhead box P3+) regulatory T-cells (Tregs) is associated with both the severity of T-cell–mediated rejection (TCMR) and the subsequent clinical prognosis. A lower Treg/Th17 infiltration ratio is significantly associated with reduced allograft function and more severe interstitial and tubular injury [26]. Also, greater infiltration of Th17 cells is significantly associated with steroid-resistant rejection, incomplete recovery, recurrent TCMR, and a lower allograft survival rate after rejection. Several mechanisms have been suggested to underlie the above. First, renal epithelial cells exposed to IL-17 produce inflammatory mediators that stimulate early alloimmune responses [13]. Second, IL-17 induces neutrophil recruitment during severe rejection, as seen in biopsies [30]. Third, Th17 cells drive alloimmune responses by promoting lymphoid neogenesis [12]. Thus, Th17 cells induce stronger and more sustained alloimmune responses, which can result in severe injury to allograft tissue.
ROLE OF TH17 CELLS IN CHRONIC KIDNEY ALLOGRAFT DYSFUNCTION
Th17 cells may be associated with severe allograft rejection. Therefore, we investigated whether Th17 cells are involved in chronic allograft dysfunction (CAD), which is the main cause of allograft failure. In human proximal renal tubular epithelial cells (HPRTEpiCs), IL-17 not only increases the production of markers of acute inflammation (IL-6 and IL-8) but also modulates the expression of profibrotic markers; e.g., CTGF (connective tissue growth factor) and ACTA-2 (α-actin 2) [31-33]. The proportion of peripheral blood Th17 cells from KT recipients with CAD was significantly increased in comparison to those of KT recipients with stable allograft function, irrespective of follow-up duration. Interestingly, the proportion of Th17 cells was higher in CAD patients than in transplant-naïve chronic kidney disease (CKD) patients, although renal function was lower in the CKD patients. This is important because uremia itself can increase the proportion of IL-17–producing effector T-cells [34]. Our results suggest that the immune response rather than renal dysfunction is responsible for the increased proportion of Th17 cells in patients with CAD (Fig. 2) [33].
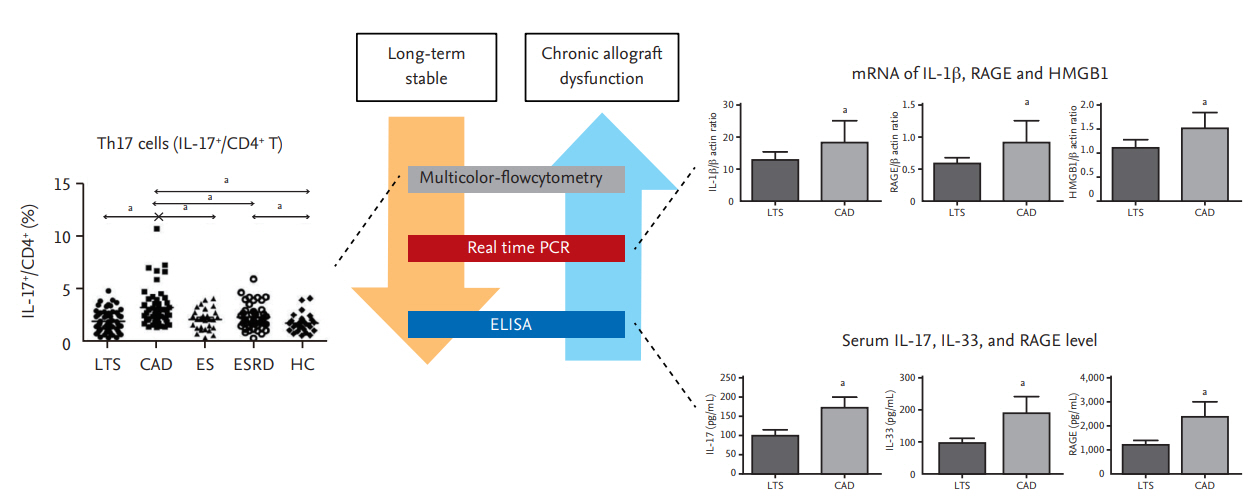
Activation of the T helper 17 (Th17) pathway in kidney transplantation recipients with chronic allograft dysfunction (CAD). The proportion of Th17 cells (interleukin 17+ [IL-17+]/cluster of differentiation 4+ [CD4+]); the IL-1β, receptor for advanced glycation end products (RAGE), and high mobility group box 1 (HMGB-1) messenger RNA (mRNA) levels; and the IL17, interleukin 33 (IL-33), and RAGE levels in peripheral blood are higher in patients with CAD. Modified from Chung et al. [41]. LTS, long-term stable; ES, early stable; ESRD, end stage renal disease; HC, healthy control; PCR, polymerase chain reaction; ELISA, enzyme-linked immunosorbent assay. ap < 0.05 for each comparison.
EFFECTS OF IMMUNOSUPPRESSIVE AGENTS ON TH17 CELLS
The incidence of early allograft loss due to acute rejection can be significantly reduced by use of immune suppressants [35]. T-cells are suppressed by treatment with a combination of tacrolimus (Tac), mycophenolate mofetil, and steroid. In addition, induction therapy using basiliximab, an anti-CD25 monoclonal antibody, suppresses the proliferation of T-cells [1]. Although maintenance immune suppression can improve the allograft survival rate in the first year after KT, there has been little improvement in the long-term outcomes [36], suggesting that the currently used immune-suppressant regimen has limitations. Tac, the major immune-suppressant used in KT recipients, blocks Th1- and Th2-associated alloimmune responses [37-39]. In contrast, few animal studies have investigated the effect of calcineurin inhibitors (CNIs) on Th17 responses. In an animal model of heart transplantation, CNI did not suppress the Th17-associated response [40]. Both Th1 and Th2 cytokines are required to reduce the IL-17 level, while administration of an anti-Th1 or -Th2 cytokine antibody increases the IL-17 level [3]. Therefore, administration of Tac may actually enhance the Th17 response. Interestingly, the percentage of Th17 cells and production of IL‑17 by effector memory T-cells (TEM) are significantly (p < 0.05) increased at 3 months after KT compared to baseline, whereas the proportions of Th1/Th2 cells and TEM cells are decreased in the early post-transplantation period [41]. In addition, Tac suppresses Th1 and Th2 cells in a concentration-dependent manner, but even a high concentration has no effect on Th17 cells in vitro [41]. This suggests that Tac-based immunosuppression may be inadequate to suppress Th17 cells in KT recipients.
STRATEGIES TARGETING TH17 CELLS
mTOR inhibitors
Activation of Th17 cells is related to more severe allograft rejection and subsequent adverse outcomes [26,42]. In addition, an increased proportion of Th17 cells in peripheral blood is associated with CAD [33]. Therefore, modulation of the Th17 response may improve allograft outcomes in KT. Mammalian target of rapamycin (mTOR) is an important regulator of helper T-cell differentiation [43-47]. Cluster of differentiation 4+ (CD4+) T-cells lacking or deficient in mTOR fail to differentiate into effector cells or Treg cells under appropriate conditions [48,49]. Also, mTOR inhibition abrogates the reprogramming of Treg cells into pathogenic Th1/Th17 effector cells [50]. Sirolimus (SRL), an mTOR inhibitor, suppresses Th17 cells in KT recipients [50,51]. Moreover, conversion from Tac to SRL inhibits the proliferation of allogeneic CD4+ T-cells and Th17 cells in vitro and ex vivo [52].
Regulation of Th17 cells by 1α,25-dihydroxyvitamin D3
Another strategy to regulate Th17 cells is to compensate Tac instead of conversion to mTOR inhibitor. Metabolic regulators, such as vitamin D, have therapeutic effects on immunologic disorders that involve the Th17 response [53-55]. Indeed, a low serum 25‑hydroxyvitamin D (25-[OH]D) level is associated with high Th17 activity in patients with autoimmune diseases or graft-versus-host disease after hematopoietic stem-cell transplantation. Furthermore, treatment with 1α,25-dihydroxyvitamin D3 (1,25[OH]2D3) ameliorates these disorders by modulating the Th17 response [53,56-58] through suppression of the mTOR/STAT3 pathway [57,59,60]. Indeed, addition of 1,25(OH)2D3 to Tac significantly inhibits Th17 cells in vitro and reduces the IL-17 and IL-22 mRNA levels in peripheral blood mononuclear cells [61]. In Jurkat cells, the mTOR/STAT3 pathway is downregulated by the addition of 1,25(OH)2D3 to Tac [61]. In an ex vivo study, treatment with 1,25(OH)2D3 for 6 months significantly decreased the Th17 level compared to baseline in 42 KT recipients [61]. Furthermore, resveratrol regulates the Th17 pathway by activating AMPK and suppressing mTOR (unpublished data). mTOR-targeted therapy suppresses Th17-related immune responses in KT recipients (Fig. 3).

Effec inhibitors on the mTOR/signal transducer and activator of transcription 3 (STAT3) signaling pathway in T helper 17 (Th17) cells. PI3K, phosphoinositide 3-kinase; S6K1, ribosomal protein S6 kinase beta-1.
CONCLUSIONS
This review summarizes the role of Th17 cells in KT. The Th17 pathway plays an important role in various types of allograft injury. Tac-based immunosuppression has a limited impact on Th17-cell-induced allograft injury. Several recent studies, including ours, suggest that mTOR-targeted strategies could suppress the Th17 pathway, but the clinical relevance is unclear. Therefore, development of strategies targeting the Th17 pathway is required to improve allograft outcomes.
Notes
No potential conflict of interest relevant to this article was reported.