Angiotensin-converting enzyme 2 and kidney diseases in the era of coronavirus disease 2019
Article information

Abstract
In the decades since the discovery of angiotensin-converting enzyme 2 (ACE2), its protective role in terms of antagonizing activation of the classical renin-angiotensin system (RAS) axis has been recognized in clinical and experimental studies on kidney and cardiovascular diseases. The effects of ACE inhibitor/angiotensin type 1 receptor blockers (ACEi/ARBs) on ACE2-angiotensin-(1-7) (Ang-(1-7))-Mas receptor (MasR) axis activation has encouraged the use of such blockers in patients with kidney and cardiovascular diseases, until the emergence of coronavirus disease 2019 (COVID-19). The previously unchallenged functions of the ACE2-Ang-(1-7)-MasR axis and ACEi/ARBs are being re-evaluated in the era of COVID-19; the hypothesis is that ACEi/ARBs may increase the risk of severe acute respiratory syndrome coronavirus 2 infection by upregulating the human ACE2 receptor expression level. In this review, we examine ACE2 molecular structure, function (as an enzyme of the RAS), and distribution. We explore the roles played by ACE2 in kidney, cardiovascular, and pulmonary diseases, highlighting studies that defined the benefits imparted when ACEi/ARBs activated the local ACE2-Ang-(1-7)-MasR axis. Finally, the question of whether ACEi/ARBs therapies should be stopped in COVID-19-infected patients will be reviewed by reference to the available evidence.
INTRODUCTION
Over the two decades since its discovery in 2000 [1,2], angiotensin-converting enzyme 2 (ACE2) has been shown to protect against certain actions of the classical renin-angiotensin system (RAS) (Fig. 1). The RAS is physiologically essential, but contributes to the pathogenesis of many diseases. The RAS is composed of an ACE, angiotensin II (Ang II) and the angiotensin type 1 receptor (AT1R). Suppression of the ACE-Ang II-AT1R axis by ACE inhibitors/AT1R blockers (ACEi/ARBs) has become the “dogma” of management for patients with kidney and cardiovascular diseases [3-6]. In contrast, the ACE2-angiotensin-(1-7) (Ang-(1-7))-Mas receptor (MasR) axis largely mitigates the consequences of AT1R activation, thus counterbalancing activation of the classical RAS [7]. Thus, blockade of the classical RAS via concurrent activation of the ACE2-Ang-(1-7)-MasR axis has emerged as an attractive therapeutic strategy [8-11]. The evidence indicates that ACEi/ARBs enhance ACE2-Ang-(1-7)-MasR axis activity [12-15], although the agents do not directly target ACE2, Ang-(1-7), or MasR, further suggesting that ACEi/ARBs are valuable for patients with kidney or cardiovascular diseases.
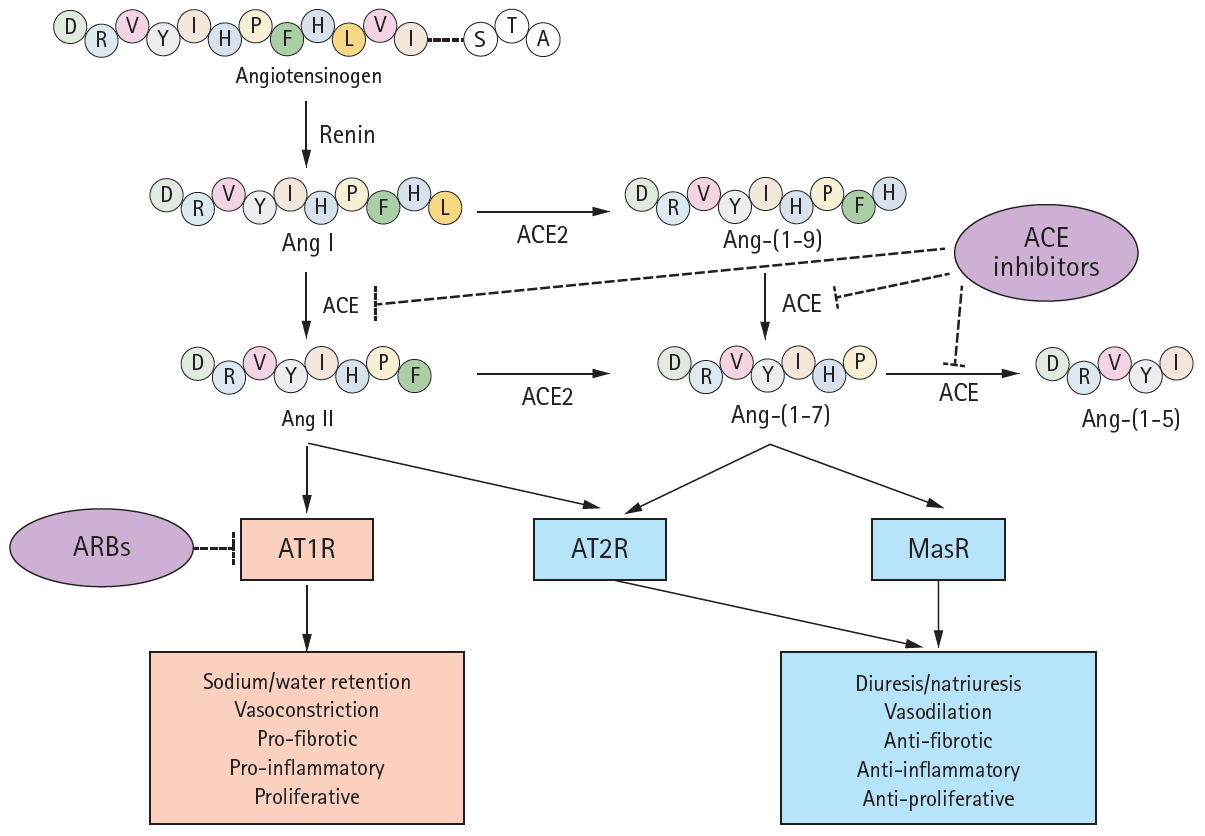
A schematic of the renin-angiotensin system (RAS) components and their modes of action. Renin converts angiotensinogen to angiotensin I (Ang I), which is subsequently cleaved by angiotensin-converting enzyme (ACE) to form Ang II. Ang II binds to its cognate G-protein-coupled receptor, angiotensin II type 1 receptor (AT1R), playing as a major effector molecule of classic RAS such as water and salt retention, vasoconstriction, and proliferative, proinflammatory, and profibrotic processes. ACE2 hydrolyzes Ang I and Ang II to Ang 1–9 and Ang-(1–7), respectively, although the enzyme efficacy for Ang II is 400-fold greater for Ang I. Angiotensin-converting enzyme 2 (ACE2) cleaves Ang II to generate Ang-(1-7), which binds to another G-protein-coupled receptor, Mas receptor (MasR). The activation of MasR is associated with abrogation of pathogenic processes medicated by AT1R, in large, counterbalances the classic RAS activation to prevent target organ damage. Ang-(1-7) is also a substrate of ACE, which is converted to an inactive metabolite. Both Ang II and Ang-(1-7) are reported to activate angiotensin II type 2 receptor (AT2R), resulting in the effect similar to MasR activation. While ACEi/angiotensin II receptor blockers (ARBs) blocks ACE and AT1R, respectively, either of ACE2, MasR, or AT2R is not inhibited by conventional inhibitors of RAS.
This dogma, however, is being threatened by the viral pandemic coronavirus disease 2019 (COVID-19). Structural studies [16-18] have shown that the causative virus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), utilizes airway epithelial ACE2 as the receptor for entry into the human host. It has been suggested that ACEi/ARBs may increase vulnerability to SARS-CoV-2 infection by upregulating the viral ACE2 receptor [19-21], triggering an intense debate as to whether ACEi/ARBs should be continued or stopped in patients with COVID-19. Indeed, clinicians have been forced to continue or discontinue ACEi/ARBs in patients with COVID-19 and underlying kidney and/or cardiovascular diseases, without reliable evidence for such decisions.
In this review, the molecular structure, function (as an enzyme of the RAS), and distribution of ACE2 will be discussed. We will explore the role played by ACE2 in kidney diseases, highlighting studies that have demonstrated the benefits afforded when ACEi/ARBs activate the local ACE2-Ang-(1-7)-MasR axis. The roles played by ACE2 in cardiovascular and pulmonary diseases will also be summarized. Finally, the differing opinions on the use or disuse of ACEi/ARBs in patients with COVID-19 will be reviewed by drawing on the evidence that has accumulated to date; decision-making must be reasonable.
ACE2 STRUCTURE, FUNCTION, AND DISTRIBUTION
More than a century after the discovery of renin, ACE2 was near-simultaneously discovered by two research groups in 2000 [1,2]. Its enzymic properties were soon established [22] and the ACE2-Ang-(1-7)-MasR axis defined [23]. The human ACE2 gene is located in chromosome Xp22 and is 40 kb in size. The gene features 18 exons, most of which resemble those of ACE [2]. As is true of ACE, the N-terminus of ACE2 is a zinc metalloprotease domain exposed to the extracellular surface. Structurally, ACE exhibits two enzymatically active sites, whereas ACE2 has only one (Fig. 2). A major difference between ACE and ACE2 lies in the distinct substrate specificities of the N-terminal domains. ACE cleaves C-terminal dipeptide residues from susceptible substrates (and is thus a peptidyl dipeptidase), converting Ang I to Ang II [22,24] and bradykinin to inactive metabolites [25]. Ang-(1-7) is also a substrate of ACE, and is converted to an inactive metabolite, Ang-(1-5), by the enzyme [26,27]. In contrast, ACE2 is a simple carboxypeptidase that hydrolyzes Ang I and Ang II to Ang 1-9 and Ang-(1-7), respectively, although the affinity for Ang II is 400-fold greater than that for Ang I [24]. ACE2 does not cleave bradykinin. Conventional inhibitors of ACE, including ramipril, block the enzymatic activity, but ACE2 is insensitive to this class of agents [22]. The C-terminus of ACE2 is a transmembrane domain with a cytosolic tail lacking any similarity to ACE. This is termed the collectrin-like domain, being a homolog of collectrin, a protein expressed in the kidney, and regulates the trafficking of amino acid transporters to the cell surface, conferring a unique function on ACE2. Unexpectedly, ACE2 serves as the host receptor for coronavirus. Interaction between the receptor-binding domain of the viral spike protein and the protease domain of the host ACE2 was shown, in the early 2000s, to be essential for entry of the SARS-CoV virus and, more recently, SARS-CoV-2 [16,17,28,29]. Compared to SARS-CoV, several mutations in amino acid residues in the interface between SARS-CoV-2 and ACE2 are evident; some may strengthen the interactions between SARS-CoV-2 and ACE2 but others may reduce the affinity by negatively affecting hydrophobic interactions and salt bridge formation [17], although the overall binding affinity of SARS-CoV-2 to ACE2 is 10- to 20-fold higher than that of SARS-CoV [29], which may partly explain the persistence of COVID-19.
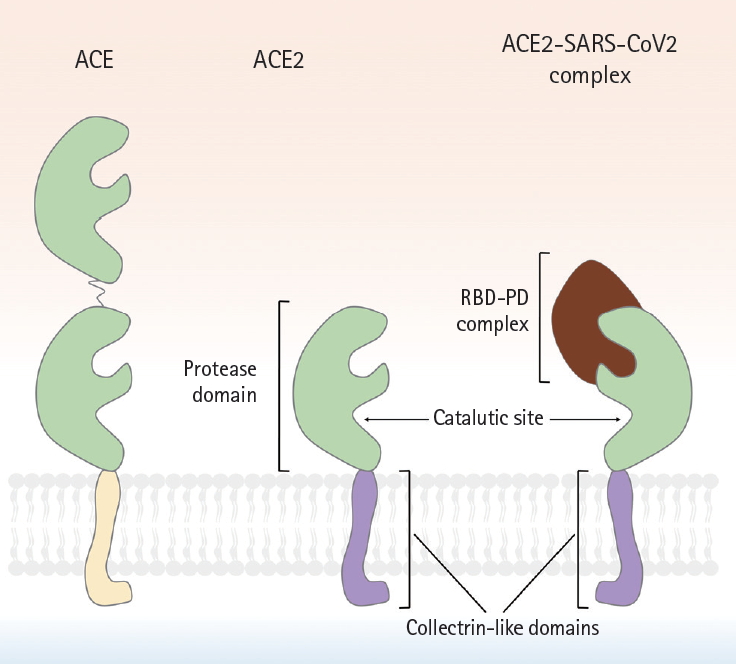
A schematic showing the molecular structures of angiotensin-converting enzyme (ACE), angiotensin-converting enzyme 2 (ACE2), and the ACE2-severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) complex. Although ACE2 is homologous to ACE. But, ACE2 has only a single active site, whereas ACE possesses 2 enzymatically active sites. Similar to ACE, the N-terminus of ACE2 is a protease domain (PD, colored in green) that is exposed to extracellular surfaces, acting as a zinc metalloprotease. The C-terminus of ACE2 is a transmembrane domain with a cytosolic tail that has no similarity with ACE. It is referred to as collectrin-like domain (colored in purple), as it is a homolog of collectrin, a protein expressed in the kidney. The interaction between receptor binding domain of viral spike protein and PD of host ACE2 is known to be crucial for viral entry of SARS-CoV in early 2000s, and more recently, SARS-CoV-2. Compared to SARS-CoV, several mutations in amino acid residues in the interface between SARS-CoV-2 and ACE2 were reported, resulting in the increase of binding affinity. RBD, receptor binding domain.
Renin converts angiotensinogen to Ang I, which is subsequently cleaved by ACE to form Ang II (Fig. 1). Ang II is a major effector molecule of the classic RAS, and binds to its cognate G-protein-coupled receptor (GPCR), termed AT1R. As AT1R activation mediates vasoconstrictive, proliferative, proinflammatory, and profibrotic processes, receptor antagonists such as ACEi/ARBs have become the cornerstone of kidney and cardiovascular disease therapeutics [4-6,30]. ACE2 cleaves Ang II to Ang-(1-7), which binds to another GPCR (MasR) [7,23,31]. As MasR activation abrogates the pathogenic processes mediated by AT1R, the ACE2-Ang-(1-7)-MasR axis essentially counterbalances the actions of the classical RAS, preventing the organ damage that will be discussed later. Conversely, RAS blockade by ACEi/ARBs upregulates ACE2 expression, although the precise mechanism was long elusive despite robust evidence from several animal studies [15,32-34]. However, the role played by the TNF-α converting enzyme (TACE) in cleavage of the ACE2 ectodomain was then discovered [35]. Using p47phox-/- mice, in which the p47phox subunit of nicotinamide adenine dinucleotide phosphate plays a crucial role in the superoxide generation induced by Ang II, it was found that oxidative stress followed by RAS activation enhanced TACE expression/activity via phosphorylation of p38 mitogen-activated protein kinase, to cleave ACE2 from cardiomyocytes (Fig. 3). Specific deletion of Tace from the myocardium prevented ACE2 shedding despite Ang II infusion [35].
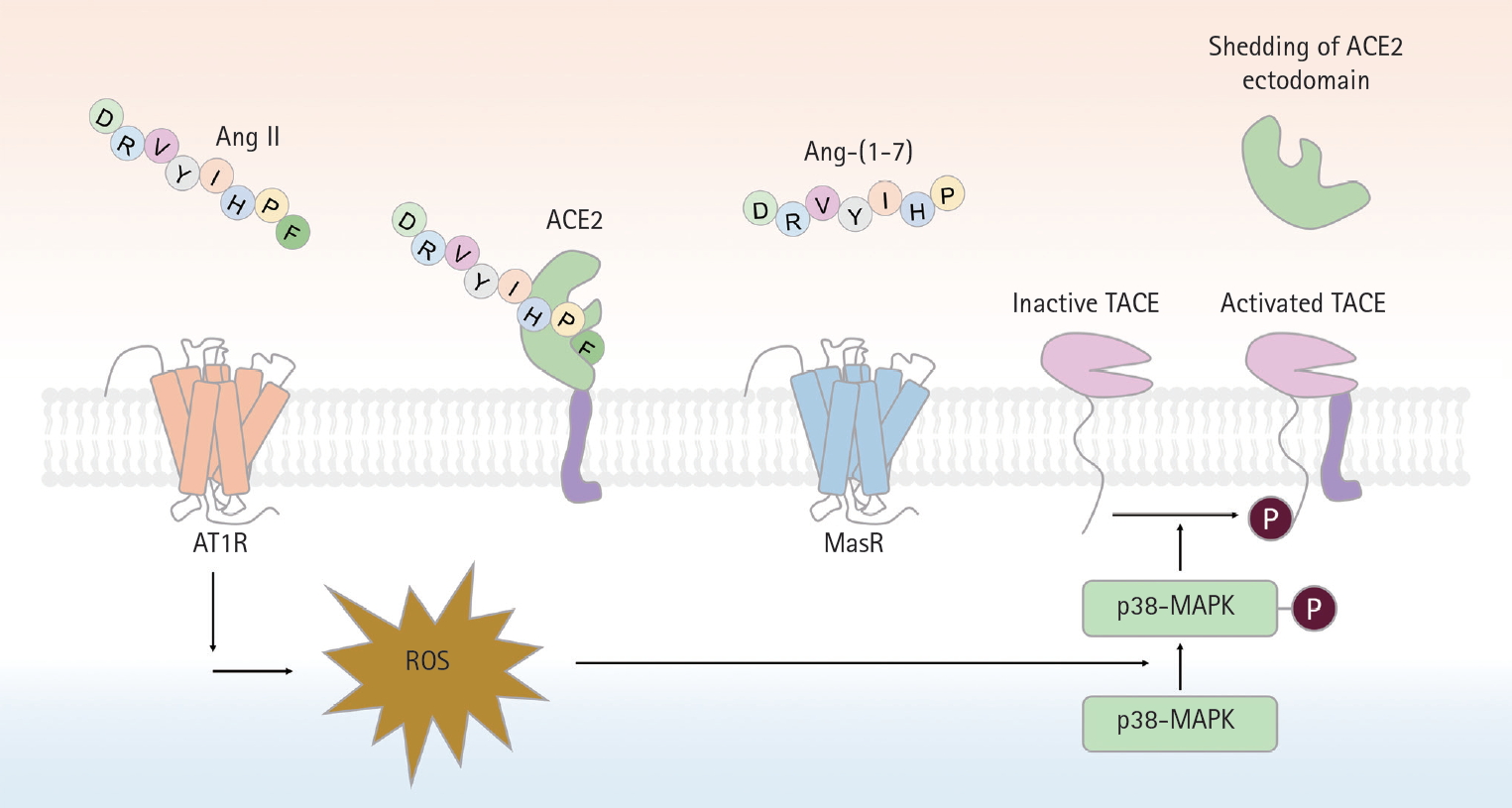
A schematic of proteolytic angiotensin-converting enzyme 2 (ACE2) ectodomain shedding after angiotensin II (Ang II)-induced TNF-α converting enzyme (TACE) activation. Activation of angiotensin II type 1 receptor (AT1R) by Ang II leads to superoxide generation, which in turn enhances phosphorylation of p38-mitogen-activated protein kinase (MAPK). Phosphorylated p38 MAPK is critical for the activation of TACE, via phosphorylation of a cytosolic residue. Activated TACE cleaved ACE2 from extracelluar surface, resuling in the shedding of ACE2 ectodomain. Ang-(1-7), angiotensin-(1-7); MasR, Mas receptor; ROS, reactive oxygen species.
The role of the angiotensin type 2 receptor (AT2R) is related to that of the ACE2-Ang-(1-7)-MasR axis, but is poorly understood (Fig. 1). AT2R is a receptor for Ang II, but the consequences of AT2R activation are opposite to those of AT1R activation. The anti-inflammatory vasoprotective effect of AT1R blockade by valsartan was significantly attenuated in AT2R knockout (KO) mice, implying that AT2R stimulation after AT1 blockade is important in terms of vascular protection [36]. Furthermore, Ang-(1-7) seems to act as an AT2R agonist, as best illustrated by the report that the anti-atherosclerotic effect of Ang-(1-7) in ApoE-/- mice was abolished by chemical inhibition of AT2R. AT2R blockade also revealed that the anti-hypertensive effect of Ang-(1-7) was largely mediated by AT2R rather than MasR [37,38]. More recently, an organ-specific protective role for ATR2 has been implied by a series of studies using a non-peptide AT2R agonist [39-42]. AT2R activation in the proximal tubules prevented sodium retention via internalization/inactivation of the major sodium transporters, and reduced the blood pressure of hypertensive rats [42]. As AT2R also exerts anti-inflammatory effects in the kidney [41], AT2R stimulation significantly ameliorated renal pathology in a rodent model of type 1 diabetes mellitus (T1DM). Thus, together with MasR, AT2R seems to be the principal receptor of an alternative, counterbalancing arm of the RAS. Organ-specific functions by the site of AT2R expression, and AT2R functions that differ from those of MasR, require further study.
Northern blotting initially implied that ACE2 expression was restricted to the heart, kidney, and testis [2]. However, later studies expanded the anatomical distribution. Lung type 2 pneumocytes and the endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) of various organs express ACE2 [43]. The epithelial cells of intestinal villi also express ACE2, which regulates amino acid transport [44]. ACE2 is normally expressed by the hepatocytes of healthy humans, and also in bile duct epithelial cells and sinusoidal ECs of patients with cirrhotic liver disease [45]. In contrast, direct evidence of ACE2 expression in the human brain remains lacking, although ACE2 expression was reported in a subset of mouse paraventricular neurons [46]. Most importantly, with the emergence of COVID-19, data based on single-cell transcriptome analysis of ACE2 expression are growing explosively [47-49], yielding many high-quality insights that will revolutionize our knowledge of ACE2 expression.
ACE2 IN KIDNEY DISEASES
Results from animal studies
The role of ACE2 in kidney diseases has been best-established in animal models of diabetic nephropathy. It is now widely accepted that intra-renal RAS activation is of particular importance in terms of the pathogenesis of such nephropathy [50]. Paradoxically, systemic RAS components are downregulated despite the robust activation of intra-renal RAS components in patients with diabetic nephropathy; this has spawned a great deal of debate [51]. T1DM induction with streptozotocin (STZ) downregulates ACE2 expression in mouse proximal tubular epithelial cells [52]. Compared to wild-type T1DM mice, deletion of Ace2 in STZ-induced T1DM mice accelerated the decline in renal function and increased the extents of glomerular and tubulointerstitial damage in a time-dependent manner [53]. Loss of Ace2 was associated with aggravated albuminuria and blood pressure elevation [52]; the responsiveness to perindopril was markedly attenuated by genetic deletion of Ace2 or treatment with an ACE2 inhibitor. Exacerbation of renal histology and albuminuria after Ace2 gene deletion has also been demonstrated in Akita mice, another animal model of T1DM [46].
The results from animal models of type 2 diabetes mellitus (T2DM) are rather complicated. Compared to wildtype mice, ACE2 expression was significantly higher in the kidney of db/db mice, a model of T2DM, with concurrent elevation of the ACE2 level in urine but not plasma [54,55]. This may reflect TACE-mediated shedding of the ACE2 ectodomain [56], which is activated by high glucose levels in various cells, including kidney, proximal tubular epithelial cells [57,58]. TACE, also known as a disintegrin and metalloproteinase 17 (ADAM17), is a metalloproteinase that can shed the ACE2 ectodomain upregulated in the kidneys of diabetic mice when its endogenous inhibitor, tissue inhibitor of metalloproteinase 3, is downregulated [55]. Together with observations from mouse models of T1DM, it has been speculated that upregulation of tubular ACE2 expression and activity may be an early event during the natural course of disease, but these features seem to decay on TACE upregulation as diabetic nephropathy progresses, although further studies are required to reveal the precise link between time-dependent glucose signaling and intra-renal regulation of ACE2. Changes in ACE2 expression have also been reported in Col4a3-/- mice, an animal model of Alport syndrome [59], characterized by a genetic defect in the glomerular basement membrane. The mice are normal at birth, but soon develop proteinuria and progressive, glomerular tubulointerstitial injuries. ACE2 expression is inversely correlated with the progression of renal injury in such mice, with a resultant rise in the Ang II level and a decline in the Ang-(1-7) level.
The effect of ACE2 administration has been examined in several animal models of kidney injury. Recombinant human ACE2 (rhACE2) effectively attenuated Ang II-mediated hypertension and renal injury [10,11], delayed progression of diabetic nephropathy in Akita mice (with a reduction in albuminuria) [60], and suppressed tubulointerstitial fibrosis in ApoE-/- animals (a model of atherosclerosis with progressive kidney lesions) by abrogating phosphorylation of AKT [61]. Recombinant murine ACE2 (rmACE2) has been given to mice with STZ-induced diabetic nephropathy, thus db/db mice and Col4a3-/-mice [62,63]. Surprisingly, a protective effect of rmACE2 was evident only in Col4a3-/- mice [62,63], regardless of the route of delivery; rmACE2 was delivered by an osmotic mini-pump in one study [62] and Ace2 minicircle DNA was injected once in the other study [63]. Thus, the differences in rmACE2 treatment efficacy may be primarily attributable to the ACE2 level/activity in the kidney tissue or urine, as urinary ACE2 activity increased significantly only in Col4a3-/- mice, despite striking rises in the serum activities of all STZ-injected mice, db/db mice, and Col4a3-/-mice [63]. One possible explanation is that rmACE2 can pass through the glomerular filtration barrier only when overt proteinuria is in play (e.g., in Col4a3-/- mice), and thus, the efficacy of systemic ACE2 delivery may be limited in mice with mild proteinuria (e.g., STZ-injected mice, db/db mice) [63].
Based on the role played by ACE2 in kidney homeostasis, several pharmacological interventions seeking to upregulate ACE2 expression have been tested in various disease models; the most consistent results are those of studies using ACEi/ARBs. One study on normotensive rats reported that lisinopril or losartan significantly augmented ACE2 activity in the renal cortex, and increased urinary excretion of Ang-(1-7) [34]. A 2-week telmisartan treatment of mice upregulated ACE2 and downregulated ACE expression in the tunica media and endothelial layer of the kidney arterioles, respectively [64]. In db/db mice given candesartan, renal tubular damage and albuminuria were ameliorated; the expression levels of ACE2, AT2R, and MasR increased; and ACE2 activity enhanced, with a reduction in extracellular signal-regulated protein kinase (ERK) 1/2 phosphorylation [12], although ultra-high doses of candesartan promoted renal injury and increased renal ERK1/2 activation. Olmesartan treatment of Col4a3-/-mice ameliorated both the glomerular and tubulointerstitial (pathological) histology, with upregulation of ACE2 expression and subsequent activation of the ACE2-Ang-(1-7)-MasR axis, despite persistence of the genetic defect [65].
Other pharmacological interventions seeking to activate the ACE2-Ang-(1-7)-MasR axis have yielded less consistent results or require further validation [66-72]. The results of treatment with diminazene aceturate (DIZE), a known ACE2 activator, are somewhat conflicting, and seem to depend on the experimental model chosen [66,67]. DIZE restored glomerular ACE2 expression and normalized whole-kidney Ang II and Ang-(1–7) levels in STZ-induced diabetic rats [66], but had no effect on blood pressure in indole-3-carbinol-induced Cyp1a1-Ren-2 transgenic rats with malignant hypertension, despite significant induction of kidney ACE2 activity and Ang-(1-7) expression [67]. This may mean that the genetic overexpression of renin was not overcome via pharmacological activation of ACE2. The fact that vitamin D is a negative regulator of renin transcription [69] has encouraged investigations of the effects of active vitamin D on kidney ACE2 expression. Calcitriol upregulated ACE2, but downregulated ACE expression in the kidney of STZ-induced diabetic rats; kidney phosphorylation of p38 and ERK was mitigated [70]. However, paricalcitol alone or in combination with aliskiren, a direct renin inhibitor, did not reduce urinary albumin excretion in non-obese diabetic mice, despite a reduction in serum ACE2 activity and enhanced cortical ACE2 expression [71]. Conversely, a role for fibroblast growth factor 23 (FGF23) in negative modulation of ACE2 expression has been suggested [73-75]. Indeed, combination FGF23/losartan compromised the effect of losartan on Ace2 mRNA upregulation in the contralateral kidney of the unilateral ureter obstruction model [72]; FGF23 alone did not affect the Ace2 mRNA level.
Together, the experimental evidence indicates that ACE2 plays a protective role in several kidney disease models, especially when activation is local rather than systemic. Pharmacological interventions enhance local ACE2 expression and activity. ACEi/ARBs have shown promising, but not fully consistent, results. Further studies on the local actions of ACE2 in kidney diseases should focus on context-dependent tailoring of therapeutics.
Results from patients with kidney diseases
Most studies on ACE2 in humans with kidney diseases are observational. In the human kidney, ACE2 has been observed in proximal tubular epithelial cells and, to a lesser degree, in glomeruli, where concurrent ACE2 downregulation and ACE upregulation have been reported in patients with T2DM and overt diabetic nephropathy [76,77], suggestive of changes in the ACE/ACE2 ratios. ACE2 downregulation in kidney tissue has been consistently reported in other studies on patients with T2DM and nodular glomerulosclerosis [78], although ACE was also downregulated; the ACE/ACE2 ratio was not measured. Analysis of urinary ACE2 protein level/activity is non-invasive and has provided valuable insights into intra-renal ACE2-Ang-(1-7)-MasR axis activity. Urinary ACE2 levels independently predict the risk of microalbuminuria, and reflect the stages and progression of chronic kidney disease (CKD) [79]. Diabetes further increases urinary ACE2 levels in such patients [80]. Intriguingly, urinary ACE2 protein excretion and activity were elevated in adolescents with uncomplicated T1DM compared to healthy controls, correlating with higher hemoglobin A1c levels, but not with the estimated glomerular filtration rate, blood pressure, or albuminuria [81], strongly suggesting that urinary ACE2 may be an early (and sensitive) biomarker of diabetic nephropathy, thus reflecting the severity of renal injury, given the mechanism of ACE2 shedding by proximal tubular epithelial cells after high-glucose exposure [56-58,82].
ACE2 IN CARDIOVASCULAR AND PULMONARY DISEASES
ACE2 in cardiovascular diseases
ACE2 is normally found in the cardiomyocytes, fibroblasts, epicardial adipocytes, and ECs of coronary vessels [35,83,84]; Ang-(1-7)/MasR is expressed in the cardiomyocytes, fibroblasts, ECs, and VSMCs of coronary vessels [85-88]. The role of the ACE2-Ang-(1-7)-MasR axis has been intensively explored in various animal models of cardiovascular disease as well as in humans. Immunohistochemistry for ACE2 in human and rat hearts revealed that ischemic injury upregulates ACE expression, principally in the vascular endothelium and smooth muscle, and less so in cardiomyocytes [89]. This seems to be a compensatory response to ischemia, rather than a mediator of tissue injury, as loss of Ace2 further accelerates maladaptive, left ventricular remodeling after myocardial infarction (MI), which was prevented by treatment with an ARB [90,91]. Overexpression of ACE2 [92] or systemic administration of Ang-(1-7) [93] also preserved cardiac function and attenuated inflammation after MI. A study of patients with idiopathic dilated cardiomyopathy revealed that heterozygotic loss of ACE2 was sufficient to promote adverse myocardial remodeling in response to pressure overload [94], implying a protective role for ACE2 in heart failure (HF). HF with a preserved ejection fraction is closely linked to obesity, and is characterized by inflammation of epicardial adipose tissue [84,95,96], which is further augmented by loss of ACE2 and increased macrophage polarization to the pro-inflammatory M1 phenotype. Ang-(1-7) attenuated M1 macrophage polarization in epicardial adipose tissue of obese Ace2 KO mice, preventing HF progression [84,97].
Activation of the ACE2-Ang-(1-7)-MasR axis assists blood pressure control. Renal Ace2 mRNA levels decreased in spontaneously hypertensive rats (SHRs) and stroke-prone SHRs [98], and lentiviral overexpression of ACE2 [99,100] or pretreatment with rhACE2 [10] attenuated blood pressure elevation in SHRs and Ang II-induced hypertensive mice, respectively. Diabetic retinopathy is another pro-inflammatory condition that is curtailed by activation of the ACE2-Ang-(1-7)-MasR axis [101,102]. Of note, ACE2 mRNA levels strongly predict microvascular disease in diabetic patients; such patients who remained free of retinopathy despite > 40 years of poor glycemic control exhibited higher levels of mRNAs transcribed from the genes of the ACE2-Ang-(1-7)-MasR axis than did age-, sex-, and glycemia-matched diabetics with retinopathy [103]. Specifically, Ang-(1-7) treatment restored the in vivo function of CD34+ bone marrow-derived vascular reparative cells and the circulating angiogenic cells that are dysfunctional in diabetics, preventing vascular injury inflicted by oxidative stress [103]. Intraocular administration of AAV-ACE2 or Ang-(1–7) reduced diabetes-induced retinal vascular leakage and inflammation, thus preventing retinopathy [102].
The efficacy of various ACEi/ARBs in terms of enhancing ACE2 expression/activity in the heart and vessels has been intensely examined, yielding quite promising and consistent results [13-15,80,104-106]. For example, olmesartan upregulated ACE2 and Ang-(1-7) expression in the aorta of SHRs [104]. Blood pressure was controlled by either lisinopril or losartan, accompanied by an increase in cardiac Ace2 mRNA levels [105]. It seems likely that the effects of ACEi/ARBs on the ACE2-Ang-(1-7)-MasR axis will further reinforce the appropriateness of ACEi/ARBs for patients with cardiovascular diseases.
ACE2 in pulmonary diseases
ACE2 is abundantly expressed by the alveolar and bronchiolar epithelium, the endothelium, and smooth muscle cells of the pulmonary vessels of rats, but not in bronchiolar smooth muscle cells [90]. ACE2 expression falls dramatically with aging in both sexes; female rats retain more ACE2 expression than do males. Acute lung injury induced by smoking downregulates lung ACE2 and upregulates ACE [107]. A precise role for ACE2 during acute lung injury has been implied by studies on Ace2 KO mice. Lung injury in such mice was induced by acid inhalation or sepsis [108], SARS-CoV infection [109], and bleomycin [110], and was more severe than in wild-type mice. rhACE2 ameliorated the histological and lung function changes in sepsis-induced [108] and pulmonary hypertension (PH) models of lung injury, and fibrosis in a bleomycin-induced model [110]. Notably, intravenous rhACE2 injection into patients with PH improved pulmonary hemodynamics and reduced the levels of oxidative and inflammatory markers [111]. Eleven patients with heritable or idiopathic PH exhibited lower ACE2 activity than healthy controls. This was the first study to explore the therapeutic efficacy of rhACE2 in humans, emphasizing the potential utility of ACE2 as a novel therapeutic. DIZE [112] and losartan [108,109,113] attenuated mouse lung injury with preservation of ACE2 expression [113], although their efficacies have not been proven in humans with pulmonary diseases.
ACEi/ARBs IN THE ERA OF COVID-19
The SARS-CoV-2 was first identified in late 2019, but lies on a continuum shared by two other highly pathogenic human coronaviruses (CoVs) described during the past two decades, thus SARS-CoV and Middle East respiratory syndrome (MERS)-CoV [114]. During the first SARS-CoV epidemic, human ACE2 was identified as the receptor for the surface spike protein (S protein) of SARS-CoV [108,109]. It remains unclear whether SARS-CoV-2 infection changes ACE2 expression; recent studies found that ACE2 expression increased after infection [115], and suggested that this was triggered by induction of interferons when the cell detected viral entry [49]. Any concern that ACEi/ARBs use might increase vulnerability to SARS-CoV by upregulating the viral ACE2 receptor was not marked prior to the emergence of COVID-19, probably because most studies on the effects of ACEi/ARBs on ACE2 expression were reported thereafter.
Initial reports indicate that comorbidities are very common in patients infected with COVID-19; these include hypertension, diabetes, coronary artery disease, and CKD [116-118]. Moreover, the frequency of pre-existing underlying conditions is considerably higher in patients exhibiting more severe clinical courses of COVID-19 infection, compared to patients with mild clinical courses [119]. Soon after identification of ACE2 as the receptor for SARS-CoV-2 [16-18], it was suggested that continued use of ACEi/ARBs in patients with underlying diseases would facilitate SARS-CoV-2 infection and increase the risk of severe disease and a fatal outcome [19]. Others later raised similar concerns [20,21,120], triggering an intense debate on continued use or cessation of ACEi/ARBs in patients with COVID-19 and underlying cardiovascular or kidney diseases. In fact, a recent letter reported that many patients from South America, Central America, and Spain, have already stopped or intend to interrupt their treatments with such drugs [121].
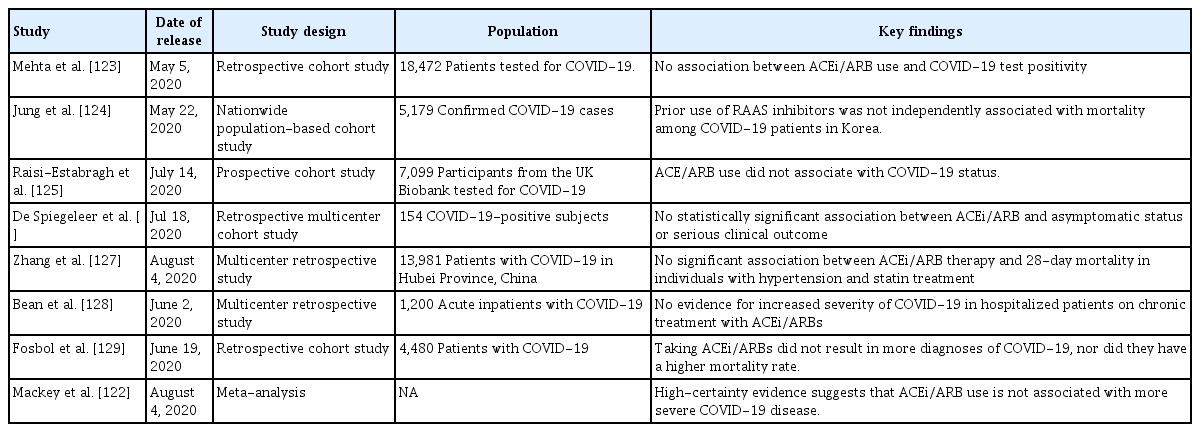
It should be emphasized; however, that, although these concerns have been (mostly) expressed by experts, they are grounded on very weak scientific evidence (Table 1) [122-129]. To date, no clinical evidence strongly recommends either the cessation or continued use of ACEi/ARBs in patients infected with COVID-19 [28,130-132]. Rather, the evidence that is available favors continued use of ACEi/ARBs in patients with COVID-19 (Table 2) [133-135], although one study suggested that ACEi/ARBs increased the risk of acute kidney injury in patients with severe COVID-19 infections [136]. For example, inhibition of AT1R by losartan attenuated SARS-CoV-induced acute lung injury in mice [108], and SARS-CoV infection downregulated ACE2 expression in mouse lungs and cultured cells, implying that uncontrolled RAS activation plays a crucial role in the pathogenesis of SARS-CoV-induced acute lung injury. One study enrolling a small number of COVID-19 patients reported that Ang II plasma levels were linearly associated with the viral load and extent of lung injury, and the authors even suggested that ARBs should be repurposed to treat COVID-19 [137]. More recently, a large retrospective multicenter study including 1,128 COVID-19 patients with hypertension on ACEi/ARBs showed that inpatient use by COVID-19 patients lowered all-cause mortality [133]. Despite potential biases attributable to extrapolation of data from mice infected with SARS-CoV, those of studies with limited numbers of patients, and those of retrospective analyses, the evidence that ACEi/ARB therapies are dangerous seems to be balanced by evidence for the opposite view. The fears are groundless. Appropriately, learned societies have recommended continuation of RAS antagonist therapies in COVID-19 patients with underlying conditions such as HF, hypertension, ischemic heart disease, or kidney disease. Unless concrete evidence supporting ACEi/ARB cessation is available; treatment should be individualized by reference to patient hemodynamic status and clinical presentation [131,132].
A summary of the results of recent studies revealing no clear association between ACEi/ARB use and COVID-19 infection status
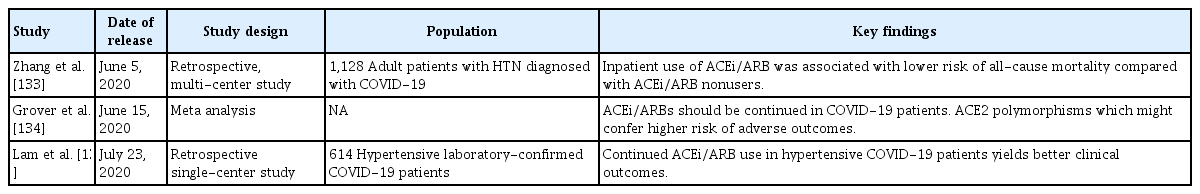
A summary of the results of recent studies revealing favorable outcomes of patients with COVID-19 infections who continued to use ACEi/ARBs
CONCLUSIONS
In the time since ACE2 was discovered, our understanding of how the RAS affects kidney and cardiovascular disease progression has expanded. Over the last two decades, the protective role played by ACE2 (in terms of antagonizing activation of the classical RAS axis) has been demonstrated in many clinical and experimental settings. The positive effects of ACEi/ARBs on ACE2-Ang-(1-7)-MasR axis activation encouraged their prescription for patients with kidney and cardiovascular diseases, until COVID-19 appeared. The human virus receptor is ACE2; the previously unchallenged utilities of ACEi/ARBs have thus been challenged. It has been suggested that ACEi/ARB use may increase the risk of SARS-CoV-2 infection by upregulation of the ACE2 receptor; however, the scientific evidence is minimal. Much accumulated evidence to date indicates that SARS-CoV-2 infection does not imply that ACEi/ARB therapy should cease in patients conventionally indicated for such drugs. Planned directed trials [122,138] will soon guide clinical decision-making in the ACEi/ARB context in COVID-19-infected patients.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by Korean government (MIST) (2020R1A2C2005620, NRF-2019R1A2C1003971, NRF-2017M3A9E8023001 & NRF-2020R1F1A1074001), and by Chonnam National University Hospital Biomedical Research Institute Grant (BCRI 20025&20076).