The 2020 revision of the guidelines for the management of myeloproliferative neoplasms
Article information

Abstract
In 2016, the World Health Organization revised the diagnostic criteria for myeloproliferative neoplasms (MPNs) based on the discovery of disease-driving genetic aberrations and extensive analysis of the clinical characteristics of patients with MPNs. Recent studies have suggested that additional somatic mutations have a clinical impact on the prognosis of patients harboring these genetic abnormalities. Treatment strategies have also advanced with the introduction of JAK inhibitors, one of which has been approved for the treatment of patients with myelofibrosis and those with hydroxyurea-resistant or intolerant polycythemia vera. Recently developed drugs aim to elicit hematologic responses, as well as symptomatic and molecular responses, and the response criteria were refined accordingly. Based on these changes, we have revised the guidelines and present the diagnosis, treatment, and risk stratification of MPNs encountered in Korea.
INTRODUCTION
Myeloproliferative neoplasms (MPNs) are clonal hematopoietic disorders characterized by the overproduction of terminally differentiated myeloid cells and an increased risk of thrombosis, bleeding, and leukemic transformation. The latest MPN classification of the World Health Organization (WHO), released in 2016, refined this disease category to include polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) as “Philadelphia-negative classical MPNs” [1].
The discovery in 2005 of JAK2V617F mutations in patients with this disease entity represented an extraordinary advancement in our understanding of the disease [2,3]. It is now well-known that both acquired and constitutive genetic alterations contribute to the pathogenesis of Philadelphia-negative MPNs. Both the annual incidence and prevalence of MPNs in Korea have increased over the years [4,5]. Given that the expected survival of the general population is increasing, MPNs are an important disease entity in Korea.
In 2015, we published the diagnostic and therapeutic guidelines for Korean patients with MPNs [6]. The revised WHO diagnostic criteria for MPNs were published in 2016 [7]. Hence, revisions of the Korean MPN guidelines are necessary to keep pace with changes in the diagnosis and treatment of the disease. Here, we have updated the epidemiology, diagnostic criteria, risk stratification, response criteria, genetic mutations, and standard treatment strategies for patients with MPNs in Korea.
EPIDEMIOLOGY
Although there are limitations to clarifying the epidemiology of MPNs because of the their indolent nature, the complexity of the diagnostic process, and partial overlap with myeloid malignancies, several studies have attempted to define the epidemiologic features of MPNs [4,5,8–10]. According to data from the Cancer Registry of Norway [10], the incidence rates of PV, ET, and PMF approximately doubled from 1995–1997 (0.4, 0.3, and 0.2 per 105 person-years, respectively) to 2010–2012 (0.7, 1.0, and 0.5 per 105 person-years, respectively). Patients with PV and ET have similar relative survival rates, whereas patients with myelofibrosis (MF) have lower relative survival, compared with the normal population [10].
In a Korean study using nationwide population-based data from the Korean National Cancer Incidence Database and the Healthcare Insurance Reimbursement and Assessment, which covers approximately 90% of the total population of Korea [4], the age-standardized incidence rates of PV, ET, and PMF in 2011 were 0.31, 0.64, and 0.11 per 105 person-years, respectively, while the respective prevalence rates were 3.28, 5.33, and 1.83 per 105 person-years. Unlike the results reported by Western studies [8,10], the incidence and prevalence of MPN in Korea increased between 2004 and 2011. The 5-year relative survival rate for all patients with MPNs during the study period was 89.3%, with the lowest rate seen in patients with MF (53.1%). Another Korean study reported similar overall outcomes [5].
In another recent big-data study in Korea [11], which evaluated 7,454 patients with MPNs who were newly diagnosed with PV, ET, or PMF from 2008 to 2016, the transformation to secondary MF or secondary acute myeloid leukemia was rare in patients with PV and ET. However, in patients with PMF, the 8-year cumulative incidence of secondary acute myeloid leukemia was 21.4%. Patients with PV or ET had an approximately 14% 8-year cumulative incidence of second primary solid tumors [11]. Consistent with the results of Western studies [12,13], Korean patients with MPNs had a twofold greater risk of developing second primary solid tumors than the general population, highlighting the importance of regular medical check-ups for malignancies in patients with MPNs.
POLYCYTHEMIA VERA
In comparing the 2016 WHO criteria [7] with the 2008 WHO criteria [14], the hemoglobin level needed for a diagnosis of PV was lowered to 16.5 g/dL in men and 16.0 g/dL in women, based on the underdiagnosis of patients with PV who had JAK2 mutations, as well as the typical clinical course of PV [15]. In addition, bone marrow examinations were more heavily emphasized and morphologic criteria were clearly described for the reproducible diagnosis of PV.
More than 90% of patients with PV harbor JAK2V617F mutations in JAK2 exon 14, while 2% to 3% of patients with PV harbor JAK2 exon 12 mutations [3,16,17]. Thus, analysis of JAK2 mutations is the most valuable laboratory test to diagnose PV. The clinical outcomes did not significantly differ between patients with JAK2V617F mutations and those with JAK2 exon 12 mutations [18]. In patients with JAK2V617F-mutated PV, a persistently high or progressive increase in the JAK2V617F allele burden was the strongest predictor of MF transformation [19].
RISK STRATIFICATION AND TREATMENT FOR PV
Although PV is classified as a neoplasm, recent studies have shown that the life expectancy of patients with PV does not differed from that of the general population [20,21]. However, symptoms and complications (e.g., pruritus, erythromelalgia, splenomegaly, thrombosis, and transformation into MF or acute myeloid leukemia) cause patients with PV to have a poor quality of life and significant morbidity. Accordingly, symptom relief and the prevention of complications and hematologic transformation are the main goals of therapy. Because of the toxicity of therapeutics, especially cytoreductive cytotoxic agents, treatment decisions should be based on a balance between side effects and risk reduction.
Unlike other MPNs, the risk stratification of PV has not changed. Age older than 60 years and a history of thrombosis have been identified as major predictors of vascular complications [22,23]. Thus, patients who had either of those two factors were defined as high-risk patients, while those who had neither were defined as low-risk patients (Table 1).

General therapeutic principles for risk stratification of patients with polycythemia vera
All patients with PV require appropriate management of cardiovascular risk factors and phlebotomy to maintain a hematocrit level of < 45% in men and < 42% in women [24,25]. The effect of phlebotomy was demonstrated in an uncontrolled study [26]. A prior randomized study, “The Intensity of CYTOreductive Therapy to Prevent Cardiovascular Events in Patients with Polycythemia Vera (CYTO-PV)”, showed that patients with a hematocrit target of < 45% had a significantly lower rate of cardiovascular death and major thrombosis than did those with a hematocrit target of 45% to 50% [27].
The efficacy and safety of low-dose aspirin (100 mg daily) in PV was verified by the European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study [28]. At the 3-year follow-up, patients receiving 100 mg aspirin showed a significant reduction in vascular events. Major bleeding was not significantly increased by aspirin. Therefore, low-dose aspirin is recommended for all patients with PV, unless contraindicated.
CYTOREDUCTIVE THERAPY FOR PV
Cytoreductive therapy using hydroxyurea or interferon-alpha (IFN-α) is indicated for high-risk patients with PV. In low-risk patients, cytoreductive therapy is recommended in the event of a progressive increase in the leukocyte and/or platelet count, severe disease-related symptoms, symptomatic splenomegaly, or phlebotomy intolerance.
Hydroxyurea is recommended as the first-line cytoreductive therapy in Korea, because IFN-α has significant toxicity and pegylated-IFN (peg-IFN) is not currently covered by the National Health Insurance system of Korea. The starting dose of hydroxyurea is 15 to 20 mg/kg/day and the dose should be adjusted for optimal count control [24,25]. Supplemental phlebotomy should be performed to maintain hematocrit at the target level.
RESISTANCE OR INTOLERANCE TO HYDROXYUREA
In 2011, European LeukemiaNet (ELN) defined the criteria for the response of patients with PV to conventional cytoreductive therapy, as well as the criteria for hydroxyurea intolerance or resistance (Table 2) [29]. In 2013, ELN and the International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) revised the response criteria to incorporate clinical, hematological, and histological assessments, and to consider disease progression and vascular events (Table 3) [30], because the previous complete response criteria were not correlated with lower thrombosis incidence or improved survival [31,32]. The revised criteria were intended to evaluate the results of clinical trials measuring the activities of drugs expected to modify the biology and natural history of PV and ET.

European LeukaemiaNet criteria for hydroxycarbamide intolerance and resistance in patients with polycythemia vera
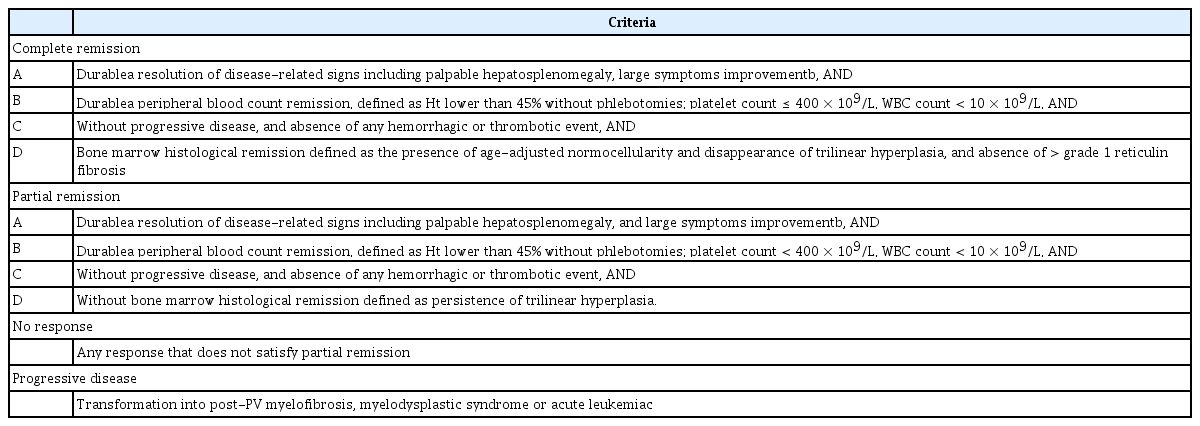
Revised (2013) European LeukemiaNet and IWG-MRT response criteria for polycythemia vera
Resistance and intolerance to hydroxyurea were observed in 5% to 10% of the patients with PV [32]. Of note, resistance to hydroxyurea was associated with a greater risk of death (hazard ratio, 5.6) and disease transformation (hazard ratio, 6.8) [32]. Therefore, we recommend bone marrow biopsy for hydroxyurea-resistant patients. Because leukocytosis and additional phlebotomy requirements, despite the use of hydroxyurea, are major thrombotic risk factors in patients with PV [33,34], a second-line drug for hydroxyurea-resistant or intolerant patients seems to be necessary.
A randomized controlled trial revealed that ruxolitinib, a JAK inhibitor, was superior to standard therapy in terms of controlling hematocrit levels, reducing spleen volume, and improving disease-related symptoms in patients with PV who had an inadequate response to or unacceptable side effects from hydroxyurea [35,36]. Peg-IFN also demonstrated efficacy in the treatment of hydroxyurea-resistant or intolerant patients with PV in a phase 2 trial, in which the overall response rate (complete or partial response) at 12 months was 60% [37]. Peg-IFN treatment was associated with a significant rate of adverse events, but most were manageable. Peg-IFN discontinuation related to adverse events occurred in only 13.9% of the patients.
In Korea, the currently available second-line therapeutics are IFN-α, peg-IFN, and ruxolitinib [35,37–39], all of which are approved by the Ministry of Food and Drug Safety. However, peg-IFN and ruxolitinib are not currently covered by the National Health Insurance system of Korea.
ESSENTIAL THROMBOCYTHEMIA
According to the 2016 WHO classification [7], bone marrow biopsy is mandatory for differentiating ET from other MPNs, especially prefibrotic PMF. Approximately 60% of patients with ET harbor JAK2V617F mutations [3,40,41]. Calreticulin (CALR) gene mutations are present in 20% to 35% of patients with ET [42,43], and a thrombopoietin receptor (MPL) gene mutation is found in 1% to 4% of patients with ET [44–46]. In the absence of any of the three major clonal mutations, testing for the most frequent accompanying mutations (e.g., ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, and SF3B1) is useful for determining the clonal nature of the disease. Although the CALR mutation is associated with a higher platelet count, lower hemoglobin level, lower leukocyte count, and lower risk of thrombosis [47–49], a large-scale study did not demonstrate that it had a significant impact on the International Prognostic Score of Thrombosis for Essential Thrombocythemia (IPSET-thrombosis) in predicting the risk of thrombosis in multivariate analysis [50]. Next-generation sequencing identified SH2B3, SF3B1, U2AF1, TP53, IDH2, and EZH2 mutations as significant risk factors for inferior overall survival (OS) and MF-free survival [51]. TP53 mutation was a predictor of inferior leukemia-free survival.
RISK STRATIFICATION AND TREATMENT OF ET
The risk stratification of ET is based on the assessment of the risk of thrombosis or bleeding complications, as in PV. However, mutational status was recently incorporated into the stratification system. In 2012, the IPSET-thrombosis was proposed, based on important factors used in assessing the risk of thrombotic complications. These factors included a prior history of venous or arterial thrombosis, age > 60 years, JAK2V617F mutation, and cardiovascular risk factors (e.g., hypertension, diabetes mellitus, and current smoking) [52]. The IPSET-thrombosis model was revised for clinical application in 2015 [53].
Table 4 describes the general therapeutic principles according to the revised IPSET-thrombosis model [54]. We recommend observation in lower (very low and low)-risk patients without cytoreductive therapy [55,56]. Low-dose aspirin in lower-risk patients is recommended in the presence of vasomotor symptoms, JAK2V617F mutation, or general indications for aspirin (e.g., cardiovascular risk factors).
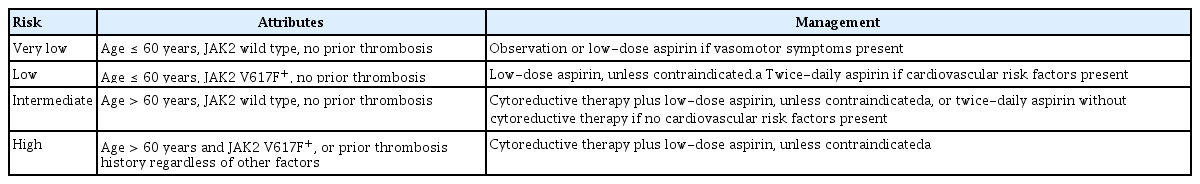
General therapeutic principles established by the 2015 revised IPSET model for thrombotic risk stratification in patients with essential thrombocythemia
Extreme thrombocytosis (i.e., platelets > 1 million/μL) may promote a hemostatic defect due to excessive adsorption of large von Willebrand factor multimers [56]. Therefore, aspirin should be avoided in patients with ristocetin cofactor activity < 30% due to the increased risk of hemorrhage [57,58]. Low-dose aspirin is acceptable if the ristocetin cofactor level is ≥ 30% [59]. Cytoreductive therapies are suggested to reduce the platelet count to 100,000 to 400,000/μL for lower-risk patients with extreme thrombocytosis [59].
In 2005, 809 patients with ET were randomly assigned to receive either hydroxyurea or anagrelide, both in combination with aspirin. Although equivalent long-term control of platelet counts was achieved in both groups, hydroxyurea plus aspirin was superior in terms of preventing both thrombosis and transformation into MF [60]. A meta-analysis also supported a favorable effect of hydroxyurea on the risk of thrombosis, major bleeding, and death (relative risk, 0.78; 95% confidence interval, 0.63 to 0.97) [61]. However, anagrelide was not inferior to hydroxyurea in preventing thrombotic complications in a subsequent trial, the ANAHYDRET Study [62]. Therefore, we recommend hydroxyurea and aspirin as first-line therapy in high-risk patients with ET, and anagrelide as second-line therapy in selected patients, including hydroxyurea-intolerant patients. We do not recommend standard IFN-α in patients with ET as a first-line treatment because of its toxicity profile, except for patients who exhibit treatment failure with hydroxyurea or who are/become pregnant during treatment.
RESISTANCE AND INTOLERANCE TO HYDROXYUREA IN PATIENTS WITH ET
Tables 5 [63] and 6 [29] depict the revised response criteria and definition of resistance and intolerance to hydroxyurea in patients with ET. A prior study revealed that approximately 10% of patients with ET became hydroxyurea-intolerant or were resistant [63]. Peg-IFN demonstrated excellent efficacy in terms of cytoreduction and the molecular response in patients with ET, without the high drug discontinuation rate observed in conventional IFN-α treatment [64–66]. This finding suggested that peg-IFN could be used in hydroxyurea-resistant or intolerant patients with ET as second-line therapy. A recent trial demonstrated the activity of peg-IFN in hydroxyurea-resistant or intolerant patients with ET [37].
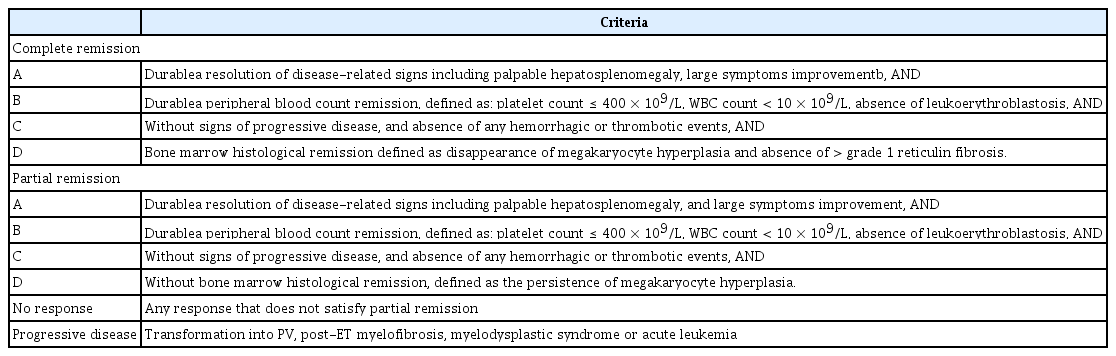
Revised (2013) European LeukemiaNet and IWG-MRT revised response criteria for essential thrombocythemia

European LeukaemiaNet criteria for hydroxyurea intolerance and resistance in patients with essential thrombocythemia
In contrast to a prospective trial of patients with PV [35,39], ruxolitinib did not demonstrate superior efficacy to the conventional, best-available therapy in hydroxyurea-resistant or intolerant patients with ET [67]. Therefore, we currently do not recommend ruxolitinib over other available drugs for those patients.
PREGNANT WOMEN AND THOSE WHO DESIRE TO BECOME PREGNANT
ET is the most common MPN in women of childbearing age [68,69], and is associated with an increased risk of both maternal and fetomaternal thrombotic complications, especially when patients have JAK2 mutations [68]. Currently, no standard approach for managing the platelet count has been established for pregnant patients with ET.
We recommend observation in lower-risk patients without a previous history of complications during pregnancy without specific therapy. The use of a platelet-lowering agent may be necessary for high-risk women with a previous history of thrombotic complications during pregnancy. Currently, both hydroxyurea and anagrelide are contraindicated for use during pregnancy. The only drug with proven safety and cytoreduction effects in pregnant patients is standard IFN-α [70–73].
A recent meta-analysis reported that the live birth rate was higher in pregnant women with MPNs who received low-dose aspirin during pregnancy than in those managed with observation alone (odds ratio, 9.48; 95% confidence interval, 4.41 to 20.41) [74]. Low-molecular-weight heparin may reduce the risk of venous thromboembolism in the antepartum and postpartum periods without increasing the risk of bleeding, although the venous thromboembolism risk was not significantly different between pregnant patients with ET who used low-molecular-weight heparin and those who did not [75].
PREFIBROTIC (EARLY STAGE) PRIMARY MYELOFIBROSIS
The 2016 WHO classification defined prefibrotic/early stage PMF (pre-PMF) [7]. Previously, PMF had been diagnosed as ET according to the 2008 WHO diagnostic criteria for MPNs, because it shares characteristics with overt PMF, such as atypical megakaryocytes, reduced erythropoiesis, high lactate dehydrogenase level, and anemia. A prior study showed that 16% of patients with ET diagnosed based on the 2006 WHO criteria had prefibrotic PMF [76]. The prognosis of patients with pre-PMF is worse than that of patients with ET in terms of OS, leukemia transformation risk, and fibrotic progression risk [76]. Therefore, differentiation between the two diseases is important. A bone marrow aspirate and biopsy with trichrome and reticulin staining are critical for differentiating ET from prefibrotic PMF [7].
The main diagnostic difference between prefibrotic and overt PMF is the grade of reticulin fibrosis in the bone marrow. Compared with overt PMF, pre-PMF causes a higher hemoglobin level and platelet count, a lower circulating blast percentage, and a lower incidence of splenomegaly. Patients with pre-PMF have a lower Dynamic International Prognostic Scoring System-plus (DIPSS-plus) risk categorization [77–79]. Differences in the distributions of ASXL1, SRSF2, U2AF1, SF3B1, EZH2, and IDH1/2 mutations, and in the incidence of unfavorable karyotypes, between the two categories of disease vary among published studies [77,79]. The OS rate was significantly higher in patients with pre-PMF than in those with overt PMF, independent of the DIPSS-plus score (p = 0.03), driver mutation status (p = 0.001), ASXL1 mutation status (p = 0.008), and SRSF2 mutation status (p = 0.02). However, no significant difference in leukemia-free survival was noted between the two categories of disease (p = 0.25) [77].
No treatment guidelines have been established for patients with pre-PMF because of the absence of long-term observations and treatment validation for this disease entity. Because this disease shares the characteristics of both ET and lower-risk overt PMF, we suggest that the treatment strategy should follow the general treatment guidelines for patients with ET or PMF, depending upon the thrombosis risk and symptom burden of the individual patient, until sufficient data have accumulated concerning this entity.
PRIMARY MYELOFIBROSIS
Patients with PV or ET show near-normal life expectancies, but the median survival of patients with PMF ranges from 4 to 5.5 years. The majority of patients experience at least one of the symptoms caused by cytopenia, splenomegaly, and increased proinflammatory cytokine levels [80,81]. The Myeloproliferative Neoplasm Symptom Assessment Form total symptom score is a simple assessment tool for checking a patient’s constitutional symptoms, splenomegaly related symptoms, and quality of life at diagnosis and during the course of treatment [82,83].
The majority of patients with PMF harbor one of three driver mutations: JAK2 (58% to 66%), CALR (23% to 35%), or MPL (7% to 8%). Patients with PMF harboring CALR type1/type1-like mutations show improved median OS (8.2 to 10.3 years) compared with those harboring CALR type 2/type 2-like (3.1 years), JAK2 (4.3 years), or MPL (4.1 years) mutations [84,85]. Approximately 10% of patients with PMF are triple-negative, which is associated with worse OS and leukemia-free survival [48,86,87].
RISK STRATIFICATION AND TREATMENT OF PMF
Prognostic scoring evolved from the International Prognostic Scoring System in 2009 [88] to the DIPSS in 2010 [89], and the DIPSS-plus in 2011 [90]. The median OS is 15.4, 6.5, 2.9, and 1.3 years for low, intermediate-1, intermediate-2, and high risk patients, respectively, according to the DIPSS-plus. Recent molecular and cytogenetic studies found mutated genes in high-molecular risk (ASXL1, EZH2, SRSF2, IDH1, and IDH2) [87,91] and high-risk karyotypes (−7/7q−, −5/5q, i(17q), +8, inv(3), 12p−, 11q23, and monosomal karyotype). Therefore, the newly developed Mutation and Karyotype-Enhanced International Prognostic Scoring System 70 (MIPSS70) and MIPSS70+ version 2.0 (integrating clinical, cytogenetic, and mutation data [92,93]), and the Genetically Inspired Prognostic Scoring System (GIPSS) model (exclusively based on genetic markers [94]), were developed in 2018 (Table 7) [95]. Because next-generation sequencing has not been popular in Korea until recently, DIPSS and DIPSS-plus remain important for risk stratification in patients with PMF. The current treatment algorithm using the risk stratification in Korea is depicted in Fig. 1.
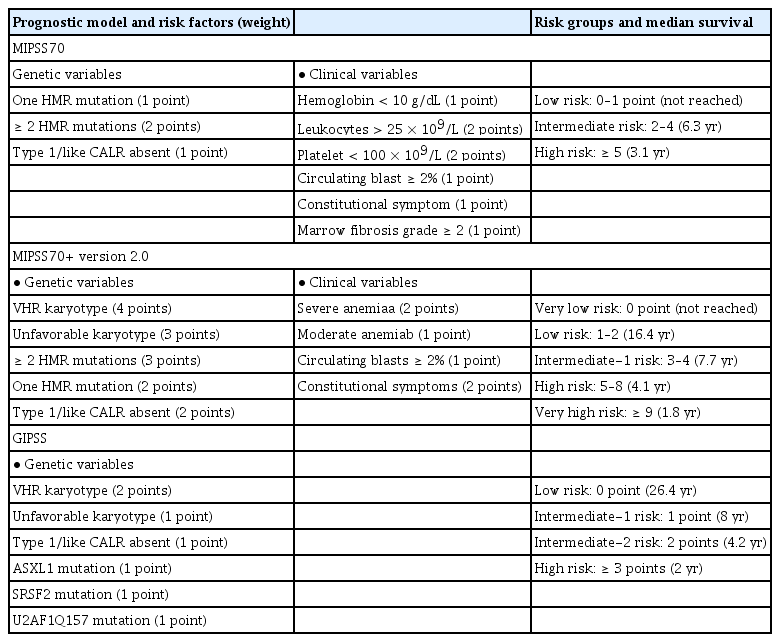
MIPSS, MIPSS-plus, and GIPSS models for primary myelofibrosis
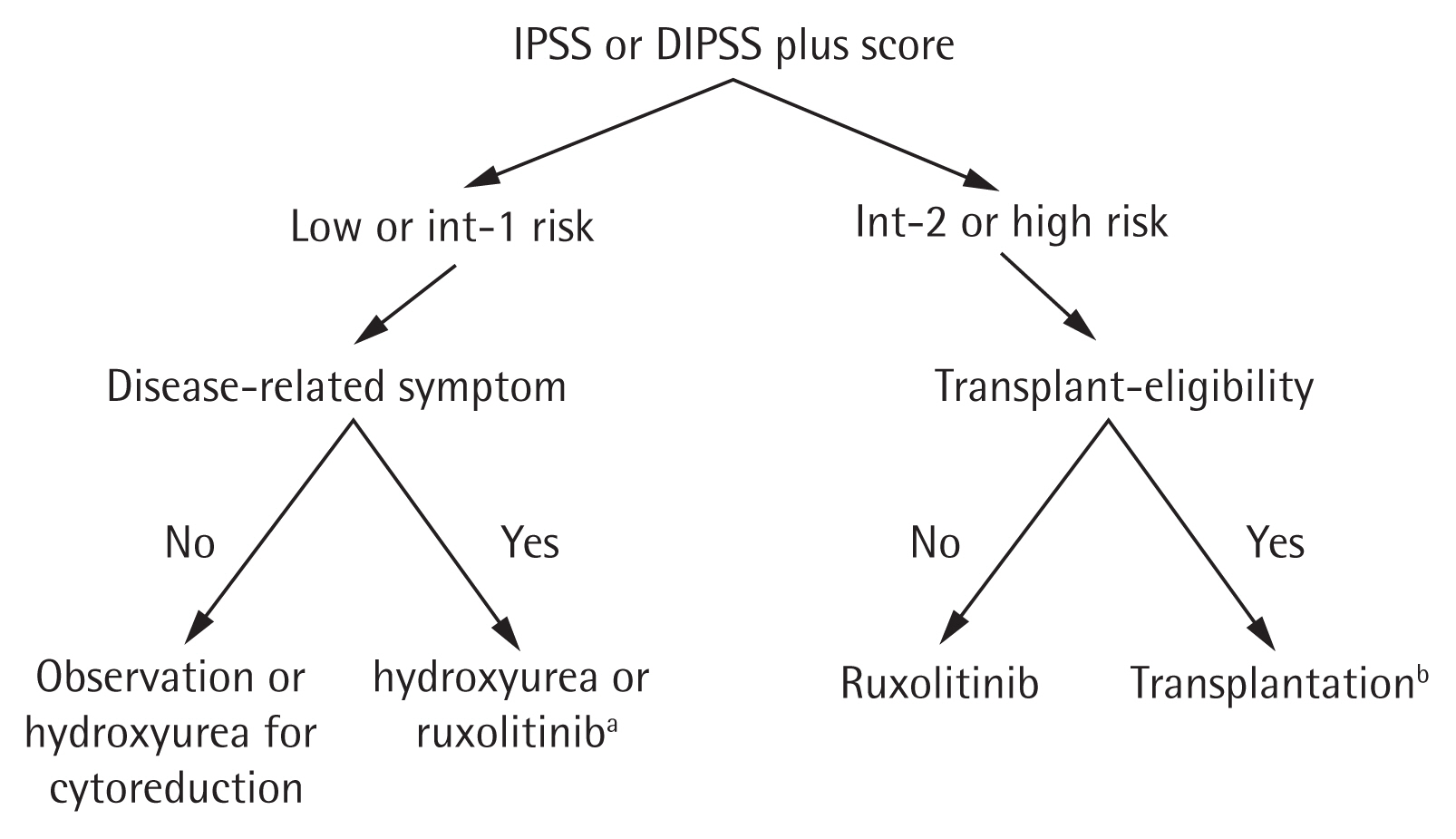
Recommended algorithm for the treatment of primary myelofibrosis. IPSS, International Prognostic Scoring System; DIPSS, Dynamic International Prognostic Scoring System. aRuxolitinib for low or intermediate-1 risk patients is approved by the Ministry of Food and Drug Safety but not currently covered by National Health Insurance system of Korea, bRuxolitinib treatment before transplantation to alleviate symptoms and splenomegaly can be considered.
TREATMENT OF SPLENOMEGALY AND CONSTITUTIONAL SYMPTOMS
Hydroxyurea can improve splenomegaly, bone pain, constitutional symptoms, and pruritus [95,96]. However, these improvements are temporary and the myelosuppressive toxicity of this agent hampers continued therapy [97,98].
Ruxolitinib was the first JAK inhibitor approved for patients with intermediate- to high-risk MF, in 2011. In two pivotal randomized trials (COMFORT-I and COMFORT-II), approximately half of the patients experienced spleen volume reductions and showed significant improvement in symptoms [99,100]. Ruxolitinib treatment also led to a significant mortality reduction (p = 0.04) and survival improvement [101]. Because grade 3–4 anemia and thrombocytopenia occurred in 45.2% and 12.9% of the patients, respectively, supportive care and dose reduction should be considered. Fedratinib [102,103], pacritinib [104], and momelotinib [105] are new JAK inhibitors that have recently shown potential for patients resistant to or intolerant to ruxolitinib.
RESPONSE EVALUATION
No drug modifying the disease activity of PMF is available. Thus, current treatment is aimed at improving anemia, reducing splenomegaly, and relieving disease-related symptoms [106]. However, recent trials using JAK inhibitors, IFNs, and other emerging drugs have attempted to demonstrate effects on molecular and cytogenetic responses, and marrow fibrosis [107–109]. Therefore, the response criteria were revised to evaluate hematologic, clinical, molecular, and cytogenetic responses (Table 8) [110].
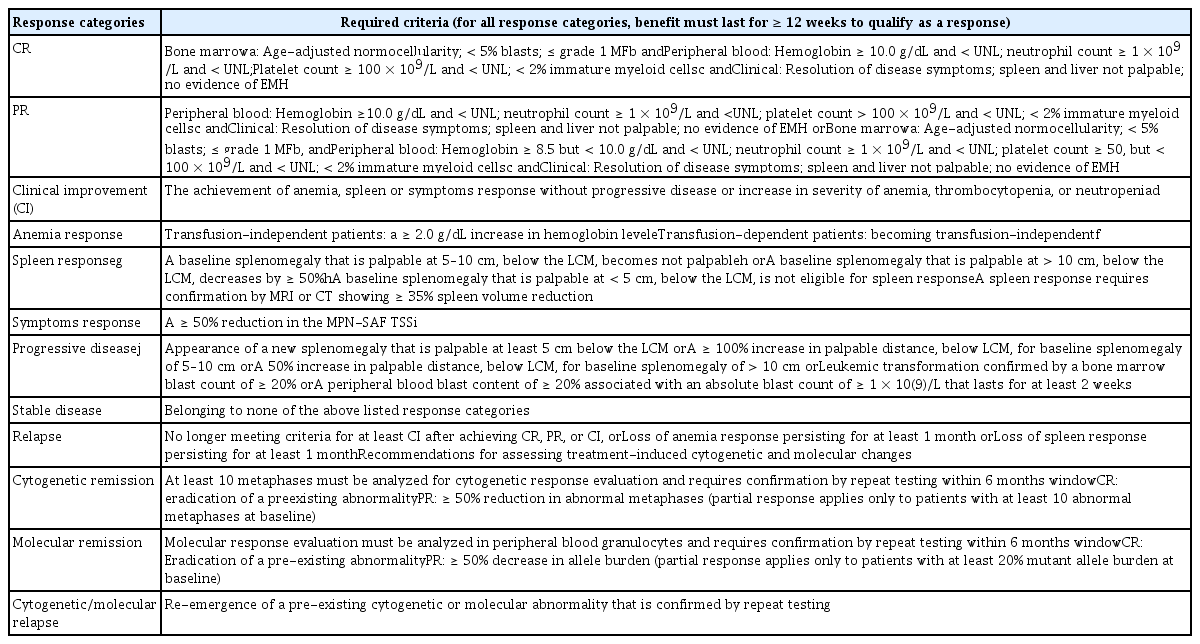
Revised (2013) IWG-MRT and European LeukaemiaNet response criteria for myelofibrosis
TREATMENT OF ANEMIA
Erythropoiesis-stimulating agents have been shown to improve anemia in 45% to 60% of MF patients. Plasma erythropoietin levels < 125 U/L have been associated with a higher probability of a response [111–113]. Androgenic steroids, such as danazol, may stimulate bone marrow function and improve hemoglobin concentrations in 30% to 40% of patients with MF [114,115]. Thalidomide [116] or lenalidomide [117], in combination with low-dose prednisone, can increase hemoglobin levels and decrease spleen size.
HEMATOPOIETIC CELL TRANSPLANTATION IN PMF
Despite the advent of JAK inhibitors, allogeneic hematopoietic cell transplantation remains the only curative treatment for PMF. Given that the median survival time of transplanted patients with PMF exceeded that of patients with PMF who did not receive transplantation in the high and intermediate-2 risk categories [118–120], allogeneic hematopoietic cell transplantation is recommended in patients with an intermediate-2 or high-risk classification, according to the DIPSS or DIPSS-plus at diagnosis or during follow-up [29,121–124]. For patients with intermediate-1 risk classification, individual counseling is necessary and we recommend MIPSS70 or GIPSS be used to assess the need for transplantation.
The Myelofibrosis Transplant Scoring System (MTSS) was suggested as a prognostic score for predicting the outcome of MF patients undergoing allogeneic hematopoietic cell transplantation based on clinical, molecular, and transplant-specific information [125]. The MTSS stratifies patients into four 5-year OS risk categories: low (85%), intermediate (64%), high (37%), or very high (22%).
The pre-transplant use of ruxolitinib may improve transplant outcomes by improving splenomegaly and performance status. Several recent trials have demonstrated the potential benefit of this strategy [126–128].
CONCLUSIONS
During the past decade, extensive knowledge concerning BCR-ABL-negative MPN has been accumulated through the detection of molecular abnormalities and many clinical analyses of affected patients. These advances led to the revision of the diagnostic criteria for MPNs by the WHO in 2016. The main change in the diagnosis was the separation of pre-PMF from the disease previously categorized as ET. This new disease classification can be differentiated using standardized bone marrow morphology and peripheral blood laboratory analysis. The hemoglobin and platelet count thresholds for the diagnosis of PV and ET were lowered in the new criteria due to the underdiagnosis of these disease entities in retrospective studies.
Because of the chronicity of MPN, risk stratification for treatment decisions is necessary to avoid unnecessary adverse effects of treatment. An in-depth understanding of the molecular abnormalities of underlying MPNs, and the clinical outcomes according to mutational status, facilitated refinement of the risk stratification. Data regarding molecular abnormalities also guided the development of targeted drugs such as JAK inhibitors, which improve the survival and quality of life of selected patients with PMF, and allow for hematologic control in hydroxyurea-resistant or intolerant patients with PV. However, newly developed drugs for the treatment of BCR/ABL-negative MPNs have not yet demonstrated efficacy in terms of improving the disease course and therapy remains supportive. A newly developed IFN agent has recently been introduced. Because immunotherapy using IFN has the potential to improve the disease course, long-term clinical data are critical.
Gene expression profiling and next-generation sequencing, which are now widely available laboratory methods, can identify various additional non-driver mutations. Additional clinical data of patients harboring these additional mutations may allow the prognosis to be better defined, and could also guide the development of agents that could change the natural course of these indolent but evolving diseases.
Notes
No potential conflict of interest relevant to this article was reported.