Differences in sputum microbiota based on cure status of patients with nontuberculous mycobacterial pulmonary disease
Article information

Abstract
Background/Aims
To analyze the characteristics of the sputum microbiota of patients with nontuberculous mycobacteria pulmonary disease (NTM-PD) based on treatment status.
Methods
Twenty-eight sputum samples from 14 patients with NTM-PD, including 14 samples from the microbiologically cured group (7 at baseline and 7 during follow-up) and 14 from the treatment-refractory group (7 at baseline and 7 during follow-up) were included in this study. Bacterial microbiota was analyzed by sequencing the V3–V4 region of the 16S rRNA gene.
Results
Among the 14 patients, most had infections with Mycobacterium avium complex (n = 6), followed by Mycobacterium abscessus (n = 5); three patients exhibited mixed infection with both organisms. Alpha-diversity was higher in the cured group than in the treatment refractory group in both the baseline sputum (ACE, p = 0.005; Chao1, p = 0.010; Jackknife, p = 0.022, 0.043; Shannon, p = 0.048) and follow-up sputum (ACE, p = 0.018). Linear discriminant analysis effect size revealed that several taxa showed differential distributions based on treatment status. At the species level, Streptococcus pneumoniae, Prevotella melaninogenica, Haemophilus parahaemolyticus, Haemophilus haemolyticus, Fusobacterium nucleatum, Neisseria elongata, and Prevotella denticola were more abundant in sputum from the microbiologically cured group than in that from the refractory group (all p < 0.05).
Conclusions
In contrast to patients with treatment-refractory NTM-PD, those with stable disease without recurrence had higher microbial diversity in their sputum, including several predominant taxa.
INTRODUCTION
Nontuberculous mycobacteria (NTM) are ubiquitous organisms, and nontuberculous mycobacteria pulmonary disease (NTM-PD) is the most common clinical manifestation of NTM infections [1]. The prevalence and medical burden of NTM-PD are increasing globally [2]. Among the causative NTM species, the Mycobacterium avium complex, which is primarily composed of M. avium and Mycobacterium intracellulare, is the most common pathogen, followed by Mycobacterium abscessus in many countries [1,3]. Although some patients achieve microbiological cure easily and without recurrence after antibiotic treatment, others experience refractory disease even after several months of treatment [4–6]. However, the pathophysiological mechanism underlying this heterogeneous treatment response remains unclear.
Microbial communities (e.g., bacteria, viruses, and fungi) can impact the development of human respiratory diseases, with disruptions at the microbial–host interface influencing disease pathogenesis [7,8]. Dysbiosis or deviation from the normal microbial composition is associated with the development and progression of respiratory diseases [9]. Reduced diversity is also observed in worsened respiratory conditions such as cystic fibrosis, asthma/chronic obstructive pulmonary disease, and idiopathic pulmonary fibrosis [10–12]. Because NTM do not reside in isolation but are part of a complex milieu of microorganisms within the host lung microbiome [7,13], changes in disease status or antibiotic treatment can alter the microbiota in patients with NTM-PD, as in patients with other lung diseases.
However, to date, to the best of our knowledge, no studies have analyzed the characteristics of the sputum microbiota of patients with NTM-PD based on NTM microbiological treatment status. Therefore, in this study, we compared the bacterial microbiota of serial sputum samples obtained from patients who achieved long-term stabilization without recurrence after microbiological cure with that of patients whose disease progressed without NTM eradication despite antibiotic treatment. Our findings elucidate the role of the microbial environment in the respiratory tract in determining the disease status of patients with NTM-PD.
METHODS
Study participants
From October 2018 to December 2022, we screened patients treated with antibiotics for NTM-PD who agreed to serial sputum sample collection in accordance with the treatment course. Paired sputum samples were collected at median intervals of 6 months from seven patients with NTM-PD undergoing follow-up after achieving microbiological cure and completing treatment and from seven patients with NTM-PD in whom the disease persisted despite >12 months of antibiotic treatment. Consequently, 28 sputum samples from 14 patients with NTM-PD were included in the study: 14 samples from the microbiologically cured group (7 at baseline and 7 during follow-up) and 14 samples from the treatment refractory group (7 at baseline and 7 during follow-up) (Supplementary Fig. 1). The bacterial microbiome in all sputum samples was analyzed, and the results were compared based on treatment status. This study was conducted on a subset of individuals from the NTM Registry of Samsung Medical Center (ClinicalTrials.gov: NCT00970801; date of first registration 02/09/2009) and was approved by the Institutional Review Board of Samsung Medical Center (IRB no. 2012-05-001, the first enrollment date of eligible patients for this study: 02/02/2021). All patients provided written informed consent.
Definition of treatment outcomes
Treatment outcomes were assessed following the NTM-NET consensus [14]. Negative sputum culture conversion was defined as at least three consecutive negative sputum cultures collected at a minimum of 4-week intervals. Microbiological cure was defined as the maintenance of multiple consecutive negative cultures, without any positive cultures of the causative species from respiratory samples, starting from culture conversion and continuing until the completion of antibiotic treatment. Treatment refractory status was defined as sustained positive cultures with causative NTM species from respiratory samples after > 12 months of antibiotic treatment.
Sputum specimens
All sputum samples were collected under identical conditions and in accordance with the sputum collection protocol. To minimize contamination, sputum was collected in the morning before the patients had eaten. Patients were instructed to submit their sputum during outpatient visits, ensuring that at least 3 mL was collected in a form that was visibly distinct from saliva. They were instructed to cough deeply into a leak-proof container. The collected sputum samples were immediately retrieved by the research staff and stored at −80°C in our institution’s laboratory. Microbiome analysis was performed within 3 days.
DNA extraction from sputum and MiSeq sequencing
DNA extraction and MiSeq sequencing were conducted similarly to those described in our previous research [7]. DNA was extracted from sputum using MP Biomedicals FastDNA® Spin Kit for soil (MPbio, Santa Ana, CA, USA) according to the manufacturer’s protocol. Subsequently, the concentration and quality of the extracted DNA were measured using the Epoch™ Spectrometer (BioTek, Winooski, VT, USA). To analyze the bacterial microbiome, the V3–V4 region of the 16S rRNA gene in bacteria was amplified.
Based on the MiSeq system protocol for preparing a 16S metagenomics sequencing library, a second polymerase chain reaction (PCR) (index PCR) was performed to attach an index sequence and an Illumina sequencing adapter to the PCR product using the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA). The second PCR-completed product was purified using the QIAquick PCR purification kit (Qiagen, Valencia, CA, USA) and quantified using the Quanti-iT PicoGreen dsDNA Assay Kit (Invitrogen, Waltham, MA, USA). The quality of the final library product was measured using the Bioanalyzer 2100 (Agilent, Palo Alto, CA, USA). The library products that passed quality control were sequenced at CJ Bioscience, Inc. (Seoul, Korea) according to the manufacturer’s instructions using the MiSeq Reagent Kit v2 (500-cycles) based on the Illumina MiSeq sequencing platform (Illumina).
Sequence analysis
Microbiome profiling was performed using the 16S-based Microbial Taxonomic Profiling (MTP) platform of the EzBio-Cloud application, which uses the 16S database version PKSSU. 4.0 [13]. MTP sets were constructed by grouping these individual MTPs, and MTP sets were compared after normalization of gene copy numbers and read count. The relative abundance of sequences was compared between MTP sets using the Wilcoxon rank-sum test. Alpha-diversity was analyzed using the species richness (ACE, Chao1, Jackknife, and number of operational taxonomic units), whereas beta-diversity was evaluated using Jensen–Shannon divergence and visualized by principal component analysis. Permutational multivariate analysis of variance (PERMANOVA) was performed to analyze statistical differences in beta-diversity.
To identify differentially distributed taxa between MTP sets, linear discriminant analysis (LDA) was performed to compare the LDA effect size (LEfSe) between different groups. Taxa with LDA score > 3.00 in the LEfSe analysis were considered significant. Significance was set at p < 0.05. Hotelling’s t-test was used to compare bacterial profiles among categories.
RESULTS
Study participants
The baseline characteristics of the patients are shown in Table 1. The median age of the patients was 61 years, and most patients (85.7%) were female. A history of pulmonary tuberculosis was reported by 28.6% of the patients. The M. avium complex was the most common causative organism, followed by M. abscessus. Three patients exhibited mixed infection with both causative organisms. In the majority of patients (85.7%), a nodular bronchiectatic pattern was observed upon radiologic examination. The median time interval between the baseline and follow-up sputum samples was 6 months (interquartile range: 6–7 mo). Detailed information on antibiotic treatments is provided in Supplementary Table 1.
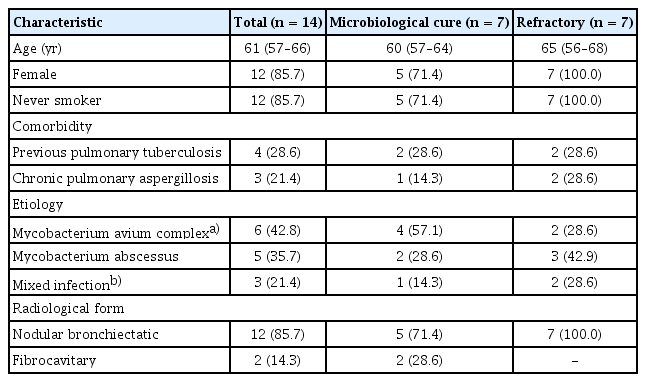
Clinical characteristics of the study patients
Comparison of microbial diversity in sputum based on treatment status
The alpha-diversity of the bacterial microbiota in the baseline sputum from the cured and refractory groups was compared (Fig. 1). The baseline sputum from the cured group had significantly higher alpha diversity and species richness than that of the treatment refractory group (ACE, p = 0.005; Chao1, p = 0.010; Jackknife, p = 0.022, p = 0.043; Shannon, p = 0.048, Fig. 1A). The follow-up sputum from the cured group also showed higher alpha-diversity than that of the treatment refractory group (ACE, p = 0.018, Fig. 1B). The total sputum from the cured group also had significantly higher alpha diversity than that of the refractory group (Supplementary Fig. 2).

Alpha-diversity of the bacterial microbiota in the sputum from the cured and refractory groups: (A) baseline samples, (B) follow-up samples. NTM, nontuberculous mycobacteria. Data are presented as box-and-whisker plots with median, interquartile range, and minimum-to-maximum values. The numbers are presented as median (interquartile range). Patients and controls were compared using the Wilcoxon’s two sample test or independent two sample t-test, as appropriate.
Beta-diversity analysis to evaluate the similarity of bacterial communities in the sputum from the two groups revealed differential species distribution in the follow-up sputum samples, but not in the baseline samples (Jensen–Shannon, PERMANOVA, p = 0.015) (Fig. 2). Differential species distribution was also observed in the total sputum from the cured and refractory groups (Jensen–Shannon, PERMANOVA, p = 0.002).
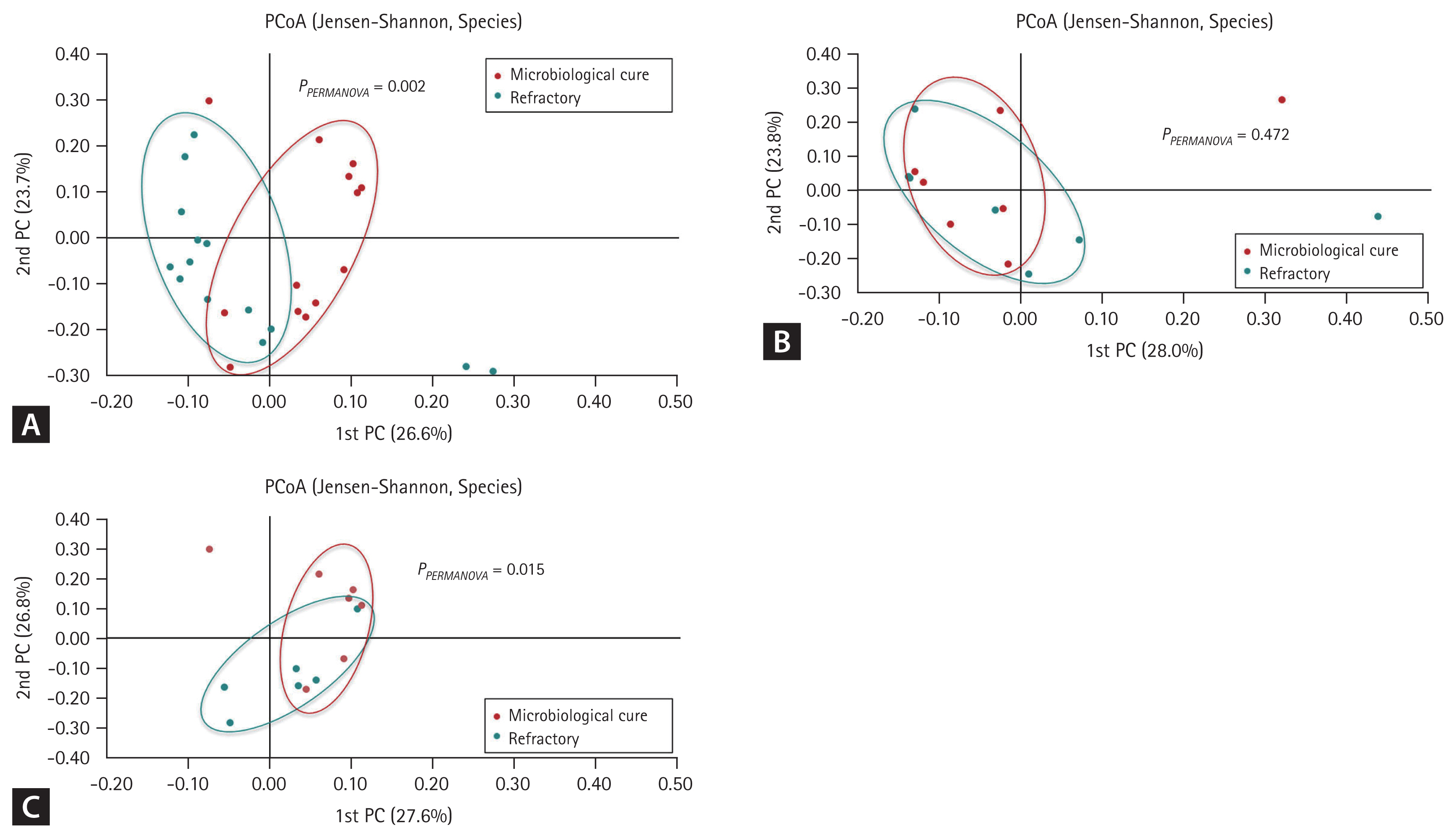
Beta-diversity of the bacterial microbiota in the study sputum from the cured and refractory groups: (A) total samples, (B) baseline samples, and (C) follow-up samples. PC, principal coordinate; PCoA, principal coordinates analysis.
Phylum- and genus-level microbial proportions according to treatment status
The proportions of microbial taxa in sputum from the cured and refractory groups were compared at the phylum and genus levels. The phyla Proteobacteria, Firmicutes, and Bacteroidetes shared dominance in the baseline sputum from both groups. In the follow-up sputum, samples from the cured group showed higher proportions of the genera Haemophilus and Campylobacter than those from the refractory group (Wilcoxon rank-sum test, p = 0.018 and 0.005, respectively) (Supplementary Fig. 3). In the total sputum from the cured and refractory groups, the genera Campylobacter and Alloprevotella were more abundant in the cured group than in the refractory group (Wilcoxon rank-sum test, p = 0.013 and 0.043, respectively) (Supplementary Fig. 3).
Analysis of species distribution differences according to treatment status
LEfSe analysis was performed to identify species that exhibited differences in distribution depending on treatment status and collection period for all sputum collected for study. Baseline and follow-up sputum samples from both groups were assigned to one of four categories to identify taxa with significant differences in distribution depending on treatment status and collection period (Table 2).

LEfSe analysis for evaluating species with distribution differences based on treatment status and collection period
Several species were more abundant in the sputum from the microbiologically cured group than in that from the refractory group, including Streptococcus pneumoniae (LDA effect = 4.890), Prevotella melaninogenica (LDA effect = 4.555), Haemophilus parahaemolyticus (LDA effect = 4.001), Haemophilus haemolyticus (LDA effect = 3.913), Fusobacterium nucleatum (LDA effect = 3.794), Neisseria elongata (LDA effect = 3.333), and Prevotella denticola (LDA effect = 3.173) (all p < 0.05; Supplementary Fig. 4).
DISCUSSION
In this study, the bacterial microbiome in sputum from patients with NTM-PD who had achieved long-term stabilization without recurrence and that from treatment refractory patients were compared. The sputum from cured patients exhibited not only high microbial diversity and richness but also several predominant taxa. Thus, a specific microbiome environment may be associated with NTM-PD disease status, and favorability of the microbial environment may contribute to disease activity in NTM-PD.
The most notable finding of our study was the prevalence of certain genera, including Streptococcus, Prevotella, Fusobacterium, Neisseria, and Haemophilus, in the sputum from patients with NTM-PD exhibiting microbiological cure and without recurrence. Studies of the lungs of healthy participants have shown that they harbor a microbiota in which the main taxa are Streptococcus, Prevotella, Fusobacterium, Haemophilus, and Veillonella [15–18], which suggests the maintenance of normal flora in the respiratory tract of stable patients without NTM-PD recurrence after treatment. In our study, the predominant bacterial species in cured patients were H. parahaemolyticus and H. haemolyticus. Although the link between the respiratory microbiome and Haemophilus remains unclear, H. haemolyticus maintains respiratory microbiome balance by competing with harmful bacteria [19]. Campylobacter and Alloprevotella were also prevalent in the sputum from cured patients with NTM-PD. Although these genera are typically present in the gastrointestinal or oral microbiota, some evidence has suggested that both are associated with pulmonary fibrosis, chronic obstructive pulmonary disease, and other respiratory diseases [20,21]. Regarding P. melaninogenica, known as a key component of the airway microbiota, a recent study has reported that the microbe is associated with reduced infection by respiratory pathogens. In a mouse model, P. melaninogenica enhanced protection against S. pneumoniae by promoting rapid pathogen clearance, and this protective effect was mediated by the recognition of P. melaninogenica lipoproteins by TLR2, the induction of TNFα, and the activity of neutrophils [22].
These findings suggest that a stable respiratory microbial environment plays a role in protecting against NTM respiratory infections. Considering that the lung microbiome exhibits distinct microbial behavior compared with microbiomes in other sites (such as the gut, skin, and vagina) [23] and that the lung microbiome maintains an ecologically dynamic state with complex fluxes of microbial immigration, emigration, clearance, and replication of local microbes [8,24], further research focusing on the microbiome in accordance with the clinical status of NTM-PD is necessary to fully comprehend the relationship with the respiratory microbiome. The precise mechanisms by which a characteristic microbiome might develop based on clinical course have not yet been elucidated in patients with NTM-PD. Additionally, no microbial taxa that exhibit antagonism against NTM have been identified to date. Normal flora or relatively low-virulence microbes may stimulate the host’s immune system, maintaining it in a “standby” state that helps prevent invasion by external pathogens. However, certainly, the results observed so far may either be causally related or could be a bystander phenomenon. Therefore, additional research is essential to analyze microbiome changes by examining more samples from patients with diverse clinical courses.
Several studies have indicated that reduced microbial diversity is correlated with increased lung disease severity, and decreased alpha-diversity has been linked to increased disease severity and bacterial pathogen colonization [25–27]. One study showed that the composition of the sputum microbiota at clinical baseline predicted the frequency of disease exacerbation in patients with bronchiectasis [28]. Consistent with those reports, our study identified high alpha-diversity in the sputum from cured patients with NTM-PD and low alpha diversity in the sputum from treatment refractory patients. Additionally, beta-diversity analysis showed compositional differences in the microbiota of both groups, with a significant difference between their follow-up samples. Thus, dysbiosis and altered microbial composition may be associated with NTM-PD progression.
Understanding the potential roles and mechanisms of the lung microbiota, particularly key functional bacteria, could offer new therapeutic targets for NTM-PD. Novel potential therapeutic approaches could involve the use of probiotics (live bacteria designed to improve health), prebiotics (food ingredients that induce specific changes in the microbiome), or antibiotics. Modifying the airway microbiota composition may range from targeting and eliminating specific strains of a single species to completely replacing the existing microbial community with a new, intact airway microbiota. However, research on the relationship between NTM and the lung microbiome is still in its early stages, and unfortunately, the current research data is insufficient to be actively applied clinically in the treatment of NTM-PD. Therefore, further research is needed.
This study has several limitations. First, the number of patients was small, and the population included patients with NTM-PD with different disease phenotypes. Differences in NTM strains and varying degrees of disease severity resulted in different airway microbiota and inflammatory signatures. Moreover, within the same individual, topographical differences in the lung microbiome were observed. Considering the limited number of patients in this study, further research involving a large patient cohort is necessary. A more comprehensive analysis, adjusting for confounding variables such as age and sex, should be conducted to examine differences among NTM subspecies, distinctions between radiological forms, and variations between initial treatment and retreatment with antibiotics. This would enhance our understanding of the microbiome characteristics in NTM-PD. Moreover, the inclusion of control groups, such as healthy adults or individuals with other respiratory diseases (e.g., asthma), which was not feasible in this study, will be essential for subsequent research. Second, several factors could influence the lung microbiota and potentially affect the results of this study, such as the host immune response, lifestyle, diet, cigarette smoking, and use of antibiotics. Unfortunately, these confounding factors were not fully considered, as they could not be controlled within the scope of this research. Third, the low alpha- and beta-diversity determined in the treatment refractory group may have been influenced by long-term antibiotic administration and NTM-PD progression. Lastly, taxonomic profiling of the microbiome based on the 16S rRNA gene does not fully reflect the population of live, metabolically active organisms.
In conclusion, our study showed that among patients with NTM-PD, patients who maintained long-term disease stabilization without recurrence exhibited higher microbial diversity in the sputum compared with treatment refractory patients. Several predominant genera were identified in samples from the cured group, including Streptococcus, Prevotella, Fusobacterium, Neisseria, and Haemophilus. A specific microbiome environment may contribute to the stabilization and cure of NTM-PD.
KEY MESSAGE
1. In patients with NTM-PD, those who maintained long-term disease stabilization without recurrence showed several dominant species distributions with higher microbial diversity compared with refractory patients.
Notes
Acknowledgments
We are grateful to the patients and researchers who participated in this study. We would also like to thank my late mentor, Professor Won-Jung Koh, for his guidance and support.
CRedit authorship contributions
Noeul Kang: data curation, formal analysis, writing - original draft, writing - review & editing; Su-Young Kim: conceptualization, methodology, data curation, formal analysis, writing - original draft, writing - review & editing; Dae Hun Kim: conceptualization, methodology, data curation, formal analysis, writing - original draft, writing - review & editing; Byung Woo Jhun: conceptualization, methodology, resources, data curation, formal analysis, writing - original draft, writing - review & editing, project administration, funding acquisition
Conflicts of interest
The authors disclose no conflicts.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2020R1C1C1008038).