


INTRODUCTION
Silicosis is a disease that affects quarry workers, stonemasons, and industrial workers who are exposed continuously to silicon particles in poorly ventilated working environments. Silicosis is a chronic pulmonary fibrotic disease that is caused by the persistent aspiration of dust containing crystalline silica. Aspirated silica can lead to cell death and tissue damage via phagocytosis by pulmonary macrophages that cannot digest these inorganic particles and thus produce reactive oxygen species (ROS) continuously. This destructive cycle results in the proliferation of fibroblasts and fibrotic scars to replace the damaged lung tissue caused by silica-induced pathologic processes [1,2].
Oxidative stress is involved in silica-induced pathogenesis by releasing ROS and may have other cellular toxic effects [3,4]. The exact mechanism by which silicosis occurs has yet to be elucidated. ROS comprises a class of ubiquitous molecules that include the superoxide anion (O2), hydrogen peroxide (H2O2), the hydroxyl radical (OH), and ROOH, all of which are produced either by normal cellular respiration processes or during the course of normal cellular signal transduction cascades such as cell growth and differentiation [5]. ROS also protects human tissues against pathogen- and foreign body-induced damage by activating powerful antiseptics [6-8]. However, in excess, ROS are toxic oxygen free radicals that cause the oxidative damage involved in aging and cell injury [9].
Peroxiredoxin (Prx), a 25-kDa enzyme first detected in yeast, belongs to a relatively new family of peroxidases and is expressed ubiquitously in all living organisms. One role for Prx is to reduce H2O2 and alkyl hydroperoxide to water (H2O) and alcohol via a catalytic system that uses thioredoxin (Trx), Trx reductase (TR), and NADPH [10,11]. Prx also plays an important role in controlling secondary signal transduction molecules by regulating low concentrations of H2O2 [5].
Although the function and expression of Prx have been reported, few studies have examined the mechanism of Prx proteolysis. Our objective herein was to determine the role of Prx in lung oxidant injuries caused by silicosis and to determine how ROS is implicated in that process. To this end, we aimed to verify the proteolytic mechanism of silicosis.
METHODS
Recombinant DNA
We amplified the complete ubiquitin cDNA sequence using polymerase chain reaction (PCR) for one cycle at 95℃ for 1 minutes, 37℃ for 1 hours, and 72℃ for 1 minutes. The human brain cDNA library (Invitrogen Life Technologies, Carlsbad, CA, USA) was used as a template from which ubiquitin was amplified using the following primers: forward, 5'-GCGGCCGCATGCAGATCTTCGTGAAGA-3', and reverse, 5'-GCGGCCGCTTACCCACCTCTGAGACGGAG-3'. The PCR conditions consisted of 30 cycles at 95℃ for 1 minutes, 55℃ for 1 minutes, and 72℃ for 1 minutes, followed by a final extension at 72℃ for 10 minutes. The amplified ubiquitin gene was ligated into the pGEM®-T Easy Vector (Promega, Madison, WI, USA) using T4 DNA ligase (Invitrogen Life Technologies, Carlsbad, CA, USA). The HA-tagged ubiquitin construct was produced by first excising the ubiquitin gene using the restriction enzymes ApaI and SalI (New England Biolabs, Beverly, MA, USA) and subsequently ligating the fragment into the pCMV-HA vector (Clontech, Mountain View, CA, USA).
Materials and methods
We used the WI26 and A549 cell lines (ATCC, Rockville, MD, USA) originating from type I and II human pulmonary epithelial cancer cells, and the Raw264.7 cell line (ATCC) originating from a mouse peritoneal macrophage cell population. The cells were cultured in the appropriate culture media as recommended by ATCC at 37℃ under 5% CO2 in 10% fetal bovine serum (Gibco BRL, Gaithersburg, MD, USA), in Dulbecco's modified Eagle's medium (Gibco BRL, Gaithersburg, MD, USA), or in RPMI1640 (Gibco BRL, Gaithersburg, MD, USA) culture medium. The culture medium was exchanged for fresh medium every 3 days. When the cells reached confluency, the cultures were trypsinized using 0.25% trypsin/ethylenediamine tetraacetic acid (EDTA; Gibco BRL, Gaithersburg, MD, USA) and divided.
In addition, we observed the expression of Prx in patients with silicosis combined with lung cancer.
Treatment of cell lines with silica and ROS blockage
In the first experiment, the A549 and WI26 cell lines were treated with 1% silica (Sigma, St. Louis, MO, USA) for 0, 24, or 48 hours and harvested for examination. In a second experiment, A549 cells were pretreated with Nacetyl-L-cysteine (NAC; Sigma, St. Louis, MO, USA) and diphenyleneiodonium (DPI; Calbiochem, Darmstadt, Germany) for 1 hours to prevent the accumulation of ROS. The cells were then treated with 1% silica, harvested, and examined at 24 and 48 hours posttreatment. In a third experiment, we added a proteosome inhibitor, 10 µM MG-132 (Calbiochem, Darmstadt, Germany), to the silica-treated cells 12 hours before we harvested the cell populations.
Transfection of HA-ubiquitin into the A549 cell line
The A549 cell cultures were established and stabilized 24 hours before they were transfected with the ubiquitin constructs. The culture medium was exchanged, and the HA-ubiquitin plasmid (20 µg) was transfected into the A549 cells (ubiquitin-A549) using Lipofectamine™ 2000 (Invitrogen Life Technologies, Carlsbad, CA, USA). After 24 hours, the transfected cells were treated with silica andMG-132.
Western blots
We extracted the total protein populations from the cell samples using protein lysis buffer (20 mM Tris [pH 7.4], 1 mM EDTA, 1 mM EGTA, 100 mM NaCl, 5 µg/mL leupeptin, 10 µg/mL aprotinin), and the extraneous debris was precipitated by centrifuging at 12,000 rpm for 10 minutes. The protein concentration in the supernatant was quan-tified using the Bradford reagent assay (Bio-Rad, Hercules, CA, USA). The proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in 12% SDS gels and then transferred to nitrocellu-lose membranes. To identify the proteins of interest, we used primary antibodies, including a polyclonal antibody against each Prx type (I-VI; Labfrontier, Seoul, Korea), a monoclonal anti-HA antibody (Sigma), and a mono-clonal anti-IκB-α antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Secondary antibodies included peroxidase-conjugated sheep anti-mouse and donkey anti-rabbit antibodies, and enhanced chemiluminescence (Amersham Biosciences, Buckinghamshire, UK) was used to detect the proteins.
Co-immunoprecipitation
To find evidence of Prx-ubiquitination, we performed a co-immunoprecipitation experiment that overexpressed the Prx isoforms or the ubiquitin protein in A549, HeLa, and Wi26 cells. We have attempted to overexpress PrxI in various cell lines. However, for unknown reason, overexpression was not successful. Therefore we have changed our strategy to overexpress ubiquitin instead and performed co-immunoprecipitation of Prx I and ubiquitin. After overexpressing ubiquitin in the aforementioned cell lines, we grew the cells in their appropriate culture media to full confluency under the aforementioned culture conditions. The cells were then harvested by trypsining (as before, with 0.25 trypsin/EDTA solution) and were lysed by adding homogenization/lysis buffer (20 mM Tris [pH 7.4], 1 mM EDTA, 1 mM EGTA, 100 mM NaCl, 5 µg/mL leupeptin, 10 µg/mL aprotinin). The lysate was centrifuged on a tabletop microcentrifuge at 4,000 rpm and only the supernatant was used for the subsequent experiment. We purified the protein samples from the A549 cells. The co-immunoprecipitation experiment was performed by adding 20 µL of protein A agarose (Upstate, Charlottesville, VA, USA), allowing it to react for 1 hours at 4℃ with gentle shaking, and separating by centrifugation at 4,000 rpm for 5 minutes. The supernatant was mixed with the anti-HA antibody (2 µg) and allowed to react for 2 hours at 4℃ with gentle shaking. Protein A agarose (20 µL) was then added to the co-immunoprecipitation samples and allowed to react for 1 hours. The immunoprecipitate was collected by centrifugation at 4,000 rpm for 5 minutes at 4℃. The pellet was washed with protein lysis buffer and centrifuged at 4,000 rpm at 4℃. This process was repeated five times to ensure purity of the precipitate. The sample buffer (1×) was added to the immunoprecipitated sample. Afterward, the resultant samples were electrophoresed (10 µL/lane) and immunoblotted with the primary antibodies.
This study was approved by the Institutional Review Board of Ajou University Hospital.
RESULTS
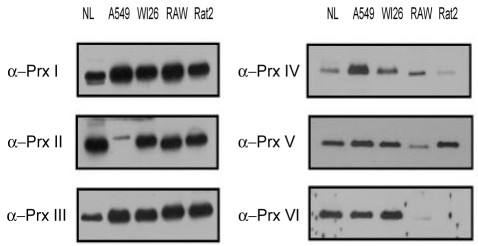
Expression of Prx isoforms in lung cells
The expression patterns of the Prx isoforms differed between the human pulmonary epithelial cell lines (A549 and WI26) and the other tested cell lines (the Raw264.7, mouse cell line; and the Rat2, rat cell line). Prx I and Prx III-VI were all expressed in the A549 cell line originating from type II human pulmonary epithelial cells, but only a low level of Prx II, an antioxidant protein in yeast and human red blood cells, was expressed. All of the Prx isoforms were differentially expressed in various cell lines in this study. Prx I and Prx III-VI were all expressed in the A549 cell with minimal expression of Prx II. All Prx isoforms were expressed in the WI26 cell line. Prx I-III and Prx V, but neither Prx IV nor Prx VI, were detected in the Raw 264.7 macrophage cell line (Fig. 1).
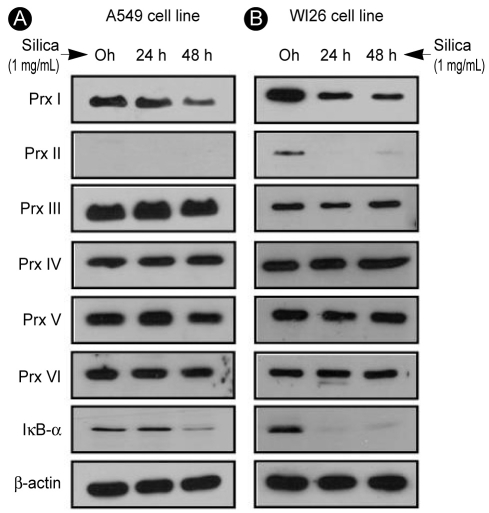
Change in Prx expression with silica treatment in Type I and II human alveolar epithelial cell lines
The A549 (type II) and WI26 (type I) cells were exposed progressively to 1% silica for 0, 24, or 48 hours, and any changes in Prx expression levels were observed and noted. In A549 cells, silica treatment caused a reduction in the expression of Prx I (Fig. 2A). In WI26 cells, silica treatment reduced the expression of Prx I and II while other Prx isoforms were intact (Fig. 2B).
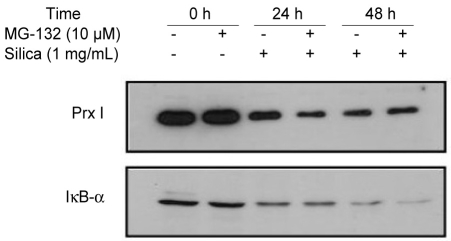
Analysis of the mechanism of silica-induced alteration of Prx expression
To confirm the relationship between the silica-induced expression of Prx and activation of ROS (silica at 1 mg/mL, 48 hours), we used NAC (1 mM, 48 hours) and DPI (1 µM, 48 hours) to remove ROS from the activated proteins linked to the oxidation-reduction (redox) reactions of the NADPH oxidase system. Silica treatment of the cell samples reduced the levels of Prx expression regardless of pretreatment with ROS inhibitors, whereas the control samples showed decreased expression of IκB-α, a com-pound that is decreased by ROS-induced protein degra-dation. Reduction of Prx expression was only dependent on the presence of silica treatment regardless of other effects of ROS (Fig. 3). Thus, it appears that the silica-induced decrease on Prx expression might not be associated with ROS but could be the result of a specific effect or direct action on the Prx protein itself.
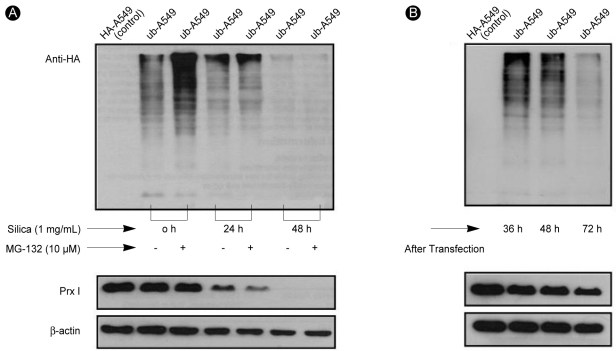
Analysis of silica-induced Prx degradation
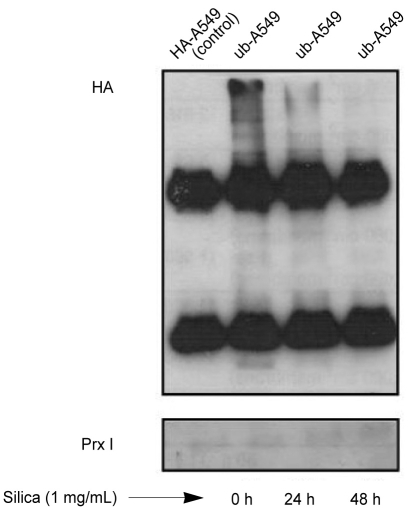
The decreased expression of Prx I induced by treatment with 1% silica in A549 cells was affected by the addition of MG-132, a potent ubiquitin-proteasomal degradation pathway inhibitor (Fig. 4). To determine whether the protease was recovered by proteasomes because of ubiquitination inactivation, we examined the amount of Prx I in the A549 cells over-expressing ubiquitin (ubiquitin-A549) by measuring the amount of Prx I bound to ubiquitin via immunoprecipi-tation. The effects of silica were not prevented by the addition of MG-132 to cells overexpressing ubiquitin (Fig. 5A), Only Lipofectamine™ 2000, which was used to obtain cells overexpressing ubiquitin, contributed to a minor decrease in the expression of Prx I (Fig. 5B). Immunoprecipitation did not detect binding between ubiquitin and Prx I indicating no ubiquitination of Prx 1 in cells (Fig. 6).
DISCUSSION
We observed decreased Prx expression levels in the A549 cells following silica treatment, regardless of the presence of ROS. However, the responsible degradation pathway was not mediated via the ubiquitin-proteasome route. ROS, including H2O2, can act as second messengers in signal transduction [12]. Excessive ROS can be detrimental to living organisms because ROS release triggers the modification of proteins, lipid membranes, chromosomes, and nucleic acids via oxidative stress, which is reportedly linked to senescence and inflammation [9].
The protein Prx, which plays a pivotal role in removing H2O2, is present ubiquitously in large amounts in every living organism from bacteria, yeast, and fungi to higher mammalian cells [6-8]. Although the exact roles of Prx in signal transduction, cellular proliferation, carcinogenesis, and protein chaperoning among living cells are unclear, the oxidation and reduction of sulfhydryl residues on the Prx protein may act as a molecular on-off signal, similar to protein phosphorylation and dephosphorylation, which serve as molecular switches of protein activation [13].
Six isoforms of Prx have been identified in mammals. Prx I and II are cytoplasmic, although small amounts of these isoforms have been detected in the nucleus. Prx III occurs only in mitochondria; Prx IV is produced in the endoplasmic reticulum and is secreted from cells; and Prx V is found either in lysosomes or in peroxisomes. Unlike the other Prx isoforms, Prx VI is an atypical subtype because it contains only one cysteine residue, and no electron donor has been identified [6,10,11].
Recent reports have helped to elucidate the role of Prx in antioxidative functions and signal transduction, as well as a possible role for this protein in the pathogenesis of many disease processes including breast cancer, skin diseases, lung cancer, radiation resistance, and RBC survival [14-20]. Most researchers have focused on the expression, function, and interactions of the Prx isoforms. However, few studies have been attempted to examine the trafficking and degradation of the Prx protein.
The goal of our study was to observe the silica-induced decrease in the expression and degradation of Prx protein levels. We found that the rate of decreased Prx expression was slower than that reported by Seo et al. [21]. This discrepancy arose presumably from differences in the characteristics of the cell lines used. Pulmonary epithelium is continually exposed to external air and thus constantly interfaces with oxidative stress components. Therefore, epithelial cells are equipped with protective mechanisms to survive under harsh oxidative stress conditions. Because the alveolar epithelium undergoes silicosis pathology, cell lines of epithelial origin such as A549 and WI26 are ideal for testing the effects of silica treatment on Prx expression. Following their examination of Prx expression levels after treating cells with MG-132, Seo et al. suggested that the proteasomal pathway is not involved in Prx degradation [21]. Herein we provide strong evidence that Prx is not ubiquitinized in pulmonary epithelial cell lines. Therefore, it is possible that Prx is degraded via the lysosomal pathway because Prx degradation did not occur via the ubiquitin-proteasomal pathway.
Our preliminary findings demonstrate that there is no decrease in Prx levels upon treatment with tumor necrosis factor alpha, which induces strong protein degradation (data not shown). This result indirectly suggests that Prx functions as a consistent and stable protein for cellular homeostasis, cellular protection, and the maintenance of the signal transduction system.
A previous study by Rhee indicated that Prx has a long half-life and coordinates closely with a regeneration system via an electrolyte transfer mechanism through Trx, TR, and NADPH [22]. The stability and consistency of Prx is further supported by evidence that when Prx is converted to peroxidated sulfinic acid, once considered an irreversible reaction, it can be reduced and regenerated by sulphiredoxin [22,23].
The release of molecules such as ROS, nitrous oxide systems, and tumor necrosis factor alpha triggered by silica inhalation can cause further injury to pulmonary macrophages, resulting in the proliferation of fibroblasts in the regeneration process [3,4,24-27]. A silica-induced decrease in Prx levels may be related to this pathogenic process. Nevertheless, silicosis pathogenesis was not fully elucidated by our experiments, and further research to determine the role(s) of Prx in this vital process is required.
Our study has several limitations. First, due to technical difficulties we did not perform microscopic observations that may verify Prx degradation. Second, we could not perform our experiments using caspase to explore apoptosis as a possible explanation for Prx degradation because the silica reacts with the culture media and renders caspase evaluations virtually impossible. However, to confirm whether silica causes apoptosis, another experiment is ongoing with other cell lines to explore relevant caspase activities. Third, we did not detect a decrease in the expression levels of Prx in the human tissue obtained from the silicosis patients, which contradicts our in vitro findings in the silica-treated A549 cells (data not shown). This conflicting observation is likely the result of using a homogenous single population of A549 cells derived from type II bronchiolar epithelial cells apart from other cell types. Tissue samples from patients harbor many heterogeneous cell types, including type I and type II alveolar epithelial cells as well as alveolar macrophages, fibroblasts, and immune competent cells, which could mask any signs of decline in Prx expression level. In support of this hypothesis is our previous obser-vation that lung cancer tissues exhibiting cellular proliferation showed a marked increase in Prx I [16]. Similarly, fibroblastic proliferation in silicotic lung tissue obtained from the surgeries reported herein may have concealed a decrease in Prx I by silica.
In conclusion, our experiments show that silica induces a decrease of Prx I expression in lung epithelial cell lines regardless of the presence of ROS, and that the silica-induced degradation of Prx does not involve the ubiquitin-proteasomal pathway.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





