


INTRODUCTION
Decreased pancreatic ╬▓-cell function and mass are core components of the progression of diabetes mellitus. Previous reports have suggested that these components are caused by reactive oxygen species, increased intracellular calcium and the activation of endoplasmic reticulum (ER) stress. These processes lead to impaired insulin secretion, decreased insulin gene expression and induction of ╬▓-cell apoptosis [1,2].
Glucotoxicity is a condition characterized by slow and progressively irreversible effects on ╬▓-cell function caused by chronic hyperglycemia. Hyperglycemia stimulates insulin secretion and activates proinsulin biosynthesis in the ERs of ╬▓-cells, and may activate ER stress [3]. Similar to chronic hyperglycemia, fatty acids become toxic when the body is chronically exposed to excessive levels; this is called lipotoxicity. In an in vitro study, when isolated islets or insulin-secreting cells were chronically exposed to elevated levels of fatty acids, glucose-induced insulin secretion [4,5] and insulin gene expression [6] decreased, whereas cell death by apoptosis [7-10] increased. There is an abundance of evidence that suggests that lipotoxicity occurs only in the presence of concomitantly elevated glucose levels [11-14] or glucotoxicity; these may be related to ER stress [15-17].
Pancreatic ╬▓-cells are highly specialized to handle the protein load within the ER. Disruption of ER homeostasis leads to the accumulation of unfolded and misfolded proteins in the ER. This condition is referred to as ER stress [18,19]. ER stress has been postulated to result from increased biosynthetic demand induced by chronic hyperglycemia and elevated free fatty acids in ╬▓-cells. This pathway is well-understood, and is based on the unfolded protein response (UPR). The UPR alleviates ER stress, restores homeostasis, and prevents cell death by inducing downstream responses that reduce new protein synthesis in the ER, increase the levels of ER chaperones to improve folding capacity and increase the capacity to dispose of misfolded proteins. If the cell is unable to successfully perform these procedures, the UPR will trigger the apoptosis cascade [20]. The three primary modulators of the UPR are inositol, which requires protein 1-╬▒, activating transcription factor 6 (ATF6) and protein kinase RNA-like ER associated kinase [21]. These sensors remain inactive via interaction with the ER chaperone BiP until activated by increased ER stress [22].
Sulfonylurea medications, which reduce blood glucose levels by stimulating insulin release from pancreatic ╬▓-cells [23], have been used for the treatment of type 2 diabetes since the early 1950s. Despite the worldwide use of sulfonylureas, the loss of ╬▓-cell mass and function caused by their use has raised concern. Studies have shown that sulfonylureas may induce apoptosis in ╬▓-cell lines and rodent islet cells [24]. The majority of patients treated long-term with sulfonylureas experience treatment failure [25]. Some evidence suggests that chronic sulfonylurea use leads to ER stress in ╬▓-cells, and this ER stress ultimately causes exhaustion of ╬▓-cell function [26]. Qian et al. [27] suggested that sulfonylureas induce the loss of ╬▓-cell function and influence the natural progression of the disease through acceleration of ER stress. Therefore, use of sulfonylureas for the treatment of type 2 diabetes mellitus may accelerate the loss of ╬▓-cell mass and function. Despite inconsistencies among studies of the effects of different types of sulfonylureas, previous results for these agents have generally been negative regarding ╬▓-cell protection [23-28]. The majority of previous studies have reported conflicting results regarding sulfonylurea treatment in ╬▓-cells without stress or with glucotoxicity only, which is quite different from the internal milieu of diabetic patients. Thus, we planned to assess the degree of apoptosis and ER stress of INS-1 cells under glucotoxic and glucolipotoxic conditions, which mimic the conditions in diabetic patients, after treatment with glibenclamide (GB).
METHODS
Cell culture
The rat insulinoma cell line INS-1 was a kind gift from Dr. Won at Yeungnam University in Korea. INS-1 cells were maintained in RPMI 1640 media (Sigma, St. Louis, MO, USA) containing 10% fetal bovine serum (FBS), 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 11 mM glucose, and 50 ┬ĄM 2-mercaptoethanol. All experiments were performed after incubation at 37Ōäā with 5% CO2 and were used between the 20th and 30th passages.
GB effects on viability and apoptosis under glucotoxic and glucolipotoxic conditions
INS-1 cells were incubated with 200 ┬ĄM palmitate and/or 33 mM glucose for 48 hours. The medium was then replaced with new medium containing 0, 10, or 100 nM GB for 24 hours. Viability was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Optical density was measured at 570 nm by an automatic plate reader. Apoptosis was assessed by annexin V staining. Cells were treated with trypsin-ethylenediaminetetraacetic acid and centrifuged at 1,500 rpm for 5 minutes at 4Ōäā. After aspirating the supernatants, cells were washed with 1-mL annexin V binding solution (140 mM NaCl, 10 mM HEPES, pH 7.4, and 2.5 mM CaCl2) and centrifuged at 1,500 rpm for 5 minutes at 4Ōäā. Supernatants were removed and 3-┬ĄL annexin V-fluorescein isothiocyanate and 10 ┬ĄL propidium iodide (PI) were added. After incubation for 15 minutes in the dark, 300 ┬ĄL fluorescence activated cell sorting (FACS) buffer (1% FBS, 0.1% NaN3) was added and analyzed by a FACSort cell sorter (BectonDickinson, BD Bioscience, San Jose, CA, USA). The percentage of apoptosis was calculated, and included annexin V positive/PI negative and both annexin V and PI positive cells. These experiments were performed in triplicate.
Changes in ER stress markers and apoptosis caused by GB treatment under glucotoxic and glucolipotoxic conditions
Markers of ER stress and apoptosis were evaluated in INS-1 cells following incubation with 10 and 100 nM GB under glucotoxic or glucolipotoxic conditions via semiquantitative reverse transcriptase-polymerase chain reaction (RT-PCR) and Western blotting. Gene expression levels of ER stress markers, including Bip-1, ATF-4, X-box binding protein-1 (XBP-1), and C/EBP-homologous protein transcription factor (CHOP), were assessed using RT-PCR. Caspase-3 and cleaved caspase-3 levels were evaluated by Western blot analysis. These experiments were also performed in triplicate.
RT-PCR
Total cellular RNA was isolated using Trizol reagent (Invitrogen, Grand Island, NY, USA). Complementary DNAs were synthesized for PCRs, which were performed using primers of ER stress markers and a premixed RT-PCR kit (Bioneer, Daejeon, Korea). The primer sequences used are listed below. Bip-1: forward, 5'-GAGATTGTTCTGGTTGGCGGATCTACTC-3'; reverse, 5'-CCATATGCTACAGCCTCATCTGGGTT-3'; ATF-4: forward, 5'-TCTGTATGAGCCCTGAGTCCTACCT-3'; reverse, 5'-GGTCATAAGGTTTGGGTCGAGAACCAC-3'; XBP-1: forward, 5'-AAACAGAGTAGCAGCACAGACTGC-3'; reverse, 5'-GGATCTCTAAGACTAGAGGCTTGGTG-3': CHOP: forward, 5'-CCTGAAAGCAGAAACCGGTC-3'; reverse, 5'-CCTCATACCAGGCTTCCAGC-3'; and GAPDH: forward, 5'-TCCCTCAAGATTGTCAGCAA-3'; reverse, 5'-AGATCCACAACGGATACATT-3'. Amplification was performed using the following conditions by the MyCycler thermal cycler (Bio-Rad, Hercules, CA, USA): predenaturation at 95Ōäā for 2 minutes, denaturation at 95Ōäā for 30 seconds, annealing at 40Ōäā for 30 seconds, extension at 72Ōäā for 30 seconds, and final extension at 72Ōäā for 7 minutes. After amplification, 5 ┬ĄL of the PCR products were subjected to electrophoresis in 1.5% agarose gels. The gels were visualized using an SL-20 DNA Image Visualizer (Seoulin Bioscience, Seongnam, Korea).
Western blot analysis
INS-1 cells were washed with phosphate-buffered saline (PBS) and lysed with a mammalian tissue lysis/extraction reagent including protease inhibitors and sodium orthovanadate. Protein quantified by the BCA protein assay kit was added to 1 ├Ś sodium dodecyl sulfate (SDS) sample buffer (50 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 50 mM dithiolthreitol, and 0.01% bromophenol blue). Proteins were separated by 12% SDS-polyacrylamide gel electrophoresis, transferred onto a polyvinylidene fluoride membrane and immunoblotted with anticaspase-3 (1 : 1,000), anticleaved caspase-3 (1 : 1,000), or anti-╬▓-actin (1 : 1,000) at 4Ōäā overnight. Secondary antibodies, such as goat antirabbit conjugated to alkaline phosphatase, were applied for 1 hour at room temperature, and membranes were developed using AP conjugate substrate kits (Bio-Rad). Developed protein bands were quantified using the Multi Gauge version 2.2 software (Fujifilm, Tokyo, Japan).
Changes in glucose-stimulated insulin secretion in INS-1 cells caused by GB treatment under glucotoxic and glucolipotoxic conditions
INS-1 cells were incubated with 10 or 100 nM GB under glucotoxic or glucolipotoxic conditions. Cells were then washed with PBS. To determine the levels of glucose-stimulated insulin secretion (GSIS), cells were starved for 5 hours in RPMI media containing 5 mM glucose and 2% FBS. Next, media were changed to KRBB solution (4.74 mM KCl, 1.19 mM KH2PO4, 1.19 mM MgCl2┬ĘH2O, 35 mM NaHCO3, and 10 mM HEPES) containing 5 or 25 mM glucose, and cells were incubated for an additional hour. Secreted insulin in the media was measured using a rat/mouse insulin enzyme-linked immunosorbent assay (ELISA) kit (Linco Research, Billerica, MA, USA).
RESULTS
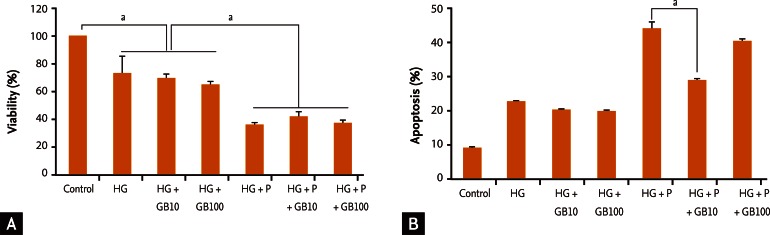
Effects of GB treatment on cell viability and apoptosis under glucotoxic and glucolipotoxic conditions
Cell viability under glucotoxic conditions decreased by more than 30% when compared to that of control cells, and decreased more than 60% under glucolipotoxic conditions. However, the addition of GB did not induce differences in cell viability (Fig. 1A).
Both glucotoxic and glucolipotoxic conditions caused apoptosis of INS-1 cells compared to control cells. The addition of GB under glucotoxic conditions did not result in significant changes in apoptosis levels. Apoptosis decreased significantly in cells grown in media containing 10 nM GB under glucolipotoxic conditions (Fig. 1B).
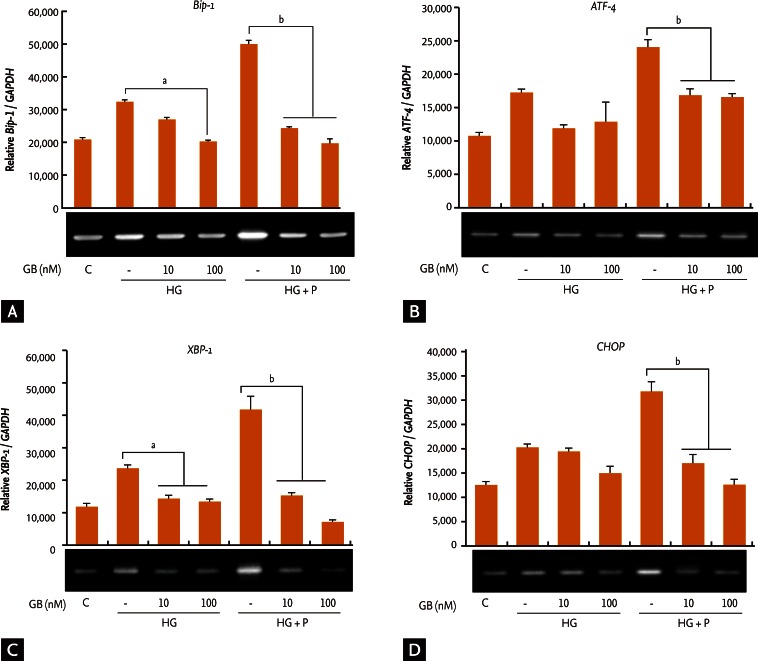
Changes in ER stress markers and apoptosis caused by GB treatment under glucotoxic and glucolipotoxic conditions
ER stress markers
Gene expression levels of ER stress markers, including Bip-1 (Fig. 2A), ATF-4 (Fig. 2B), XBP-1 (Fig. 2C), and CHOP (Fig. 2D), increased under glucotoxic and glucolipotoxic conditions when compared to the normal glucose control. GB treatment in INS-1 cells under glucotoxic conditions somewhat decreased the expression of ER stress markers Bip-1, ATF-4, XBP-1, and CHOP. The decreases in Bip-1 and XBP-1 levels were statistically significant. Under glucolipotoxic conditions, levels of all ER stress markers were reduced after the addition of GB.
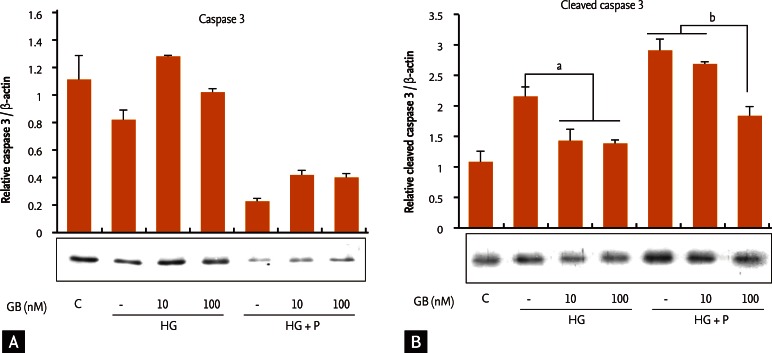
Proapoptotic markers
Caspase-3 levels decreased while those of cleaved caspase-3 increased in cells incubated under glucotoxic and glucolipotoxic conditions compared with cells incubated under normal conditions (Fig. 3). Specifically, the level of the cleaved form of caspase-3 increased compared to total caspase-3 levels. Although caspase-3 did not show a significant change after GB addition under glucotoxic or glucolipotoxic conditions, the slight increase in caspase-3 and decrease in cleaved caspase-3 levels suggest a definite decrease in the percentage of cleaved caspase-3.
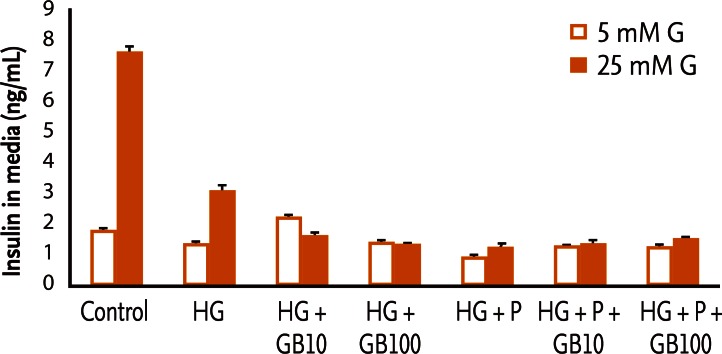
GB effects on GSIS under glucotoxic and glucolipotoxic conditions
The impairment of glucose-dependent insulin secretion caused by glucotoxicity and glucolipotoxicity did not recover after the addition of GB (Fig. 4).
DISCUSSION
Previous studies have shown that diabetic patients have increased ER stress because islets isolated from type 2 diabetics have increased numbers of ER chaperones and CHOP, along with enlarged ERs [11,12,29]. Elevated ER stress caused by high glucose and fatty acids has been suggested as one of the causes of decreased ╬▓-cell function and mass, as shown in our study, as well as in previous reports.
Sulfonylureas have been predicted to accelerate apoptosis and the dysfunction of ╬▓-cells due to their mechanism of action. Previous studies have reported inconsistent results depending on the cell type used, the concentrations and types of sulfonylureas and the duration of exposure to them. In most studies using GB, authors have found increased ╬▓-cell apoptosis and decreased ╬▓-cell function [23,28].
The effects on ╬▓-cells of GB should be verified under conditions similar to those in diabetic patients treated with GB, such as glucotoxic and glucolipotoxic conditions. The peak plasma concentrations of GB, when administered at doses of 2.5 mg, are 0.15 ┬ĄM [30]. This study was performed using lower concentrations of GB (10 or 100 nM) than those used in other reports. Apoptosis did not increase with the addition of low concentrations of GB under glucotoxic or glucolipotoxic conditions. Interestingly, apoptosis decreased significantly in media containing 10 nM GB under glucolipotoxic conditions. ER stress and proapoptotic markers decreased when low concentrations of GB were administered under glucolipotoxic conditions. These results suggest that low concentrations of GB reduce ER stress under extremely stressful conditions, such as glucotoxicity and glucolipotoxicity. Another probable hypothesis is a binary switch of ER stress [31]. The UPR has both adaptive and apoptotic effectors that depend the tolerability of ER stress. Under tolerable ER stress conditions, the UPR acts to reduce ER stress and promote ╬▓-cell survival. Although ER stress cannot be entirely responsible for the pathogenesis of type 2 diabetes, it is an important finding that apoptosis of ╬▓-cells and proapoptotic markers decrease after treatment with low concentrations of GB.
In the United Kingdom Prospective Diabetes Study, loss of ╬▓-cell function was identified in all type 2 diabetic patients. The rates of decline were identical among all treatment groups, which included sulfonylurea, metformin, and conventional treatments [32]. Thus treatment with low-dose sulfonylureas may not accelerate progression of type 2 diabetes.
Our study has several limitations: 1) only one ╬▓-cell line was used; 2) only in vitro data were obtained; and 3) a more detailed mechanism was not established. More studies using various cell lines and primary human cell cultures are needed to confirm these results. An in vivo study is also necessary. Although animal models of type 2 diabetes may mimic internal ER stress under glucotoxic or glucolipotoxic conditions, measuring the degree of toxicity is difficult. When examining an in vivo study, it is difficult to rule out interactions with other parameters. However, well-designed in vivo experiments are needed to confirm our results. This study cannot explain the mechanism by which low concentrations of GB decrease ER stress and apoptosis. The phosphatidylinositol 3-kinase (PI3K)/Akt pathway [33] and antiapoptotic markers, such as apoptosis antagonizing transcription factor [34], have recently been reported to be associated with ER stress. PI3K and Akt were tested using the same conditions as described in this study; however, they were unaffected by addition of GB under glucotoxic or glucolipotoxic conditions (data not shown). A further study should attempt to determine why GSIS did not recover after GB treatment despite the beneficial effects on apoptosis and ER stress.
Low concentrations of GB resulted in antiapoptotic effects, which were mediated by attenuation of ER stress under glucotoxic and glucolipotoxic conditions. Low-dose sulfonylurea treatment in type 2 diabetes may not directly cause secondary ╬▓-cell failure. To confirm these results, further studies using various cell lines will be needed to elucidate a more detailed mechanism.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





