


INTRODUCTION
Oxidative stress has been implicated in the pathogenesis of various diseases, including atherosclerosis, fatty liver disease, cancer, and neurodegenerative diseases [1,2]. There is now abundant experimental evidence that oxidation of biological molecules such as lipids, proteins, and DNA, mediated by reactive oxygen species (ROS) and free radicals, results in damage to biological membranes, modification of proteins, deactivation of enzymes, and modification of DNA. Consequently, the role of antioxidants against oxidative stress in the prevention and treatment of diseases has received much attention not only from scientists but also from the general public [3]. It is expected that, if oxidative stress plays a causative role, then antioxidants should reduce the risk or be useful in the prevention and treatment of these diseases.
Several epidemiological studies carried out in the 1980s suggested beneficial roles of antioxidants such as vitamin E and carotenoids. The consumption of fresh fruits and green-yellow vegetables is inversely related to the incidence of cancer [4]. This is attributed, at least in part, to the numerous phytochemical compounds in plant foods, many of which are potent antioxidants. The Multinational Monitoring of Trends and Determinants in Cardiovascular Disease (MONICA) study demonstrated an inverse association between vitamin E level in plasma and mortality attributable to ischemic heart disease and cancer [5]. Strongly positive findings in several large cohort studies have supported a protective role for antioxidants. Enthusiasm has grown to the point that dietary supplements containing antioxidants are very popular among a large portion of the population.
However, large-scale randomized clinical trials and meta-analyses have reported disappointing and conflicting results on the effects of vitamin E. Not only have there been several null findings, but some studies have reported that intake of large doses of vitamin E may be harmful, which has resulted in much debate [6-8].
This article reviews the roles and effects of vitamin E and addresses the following problems considering the available scientific evidence. Do free radicals cause oxidative damage and associated diseases? Does vitamin E prevent oxidation mediated by free radicals, and associated diseases? Why are the results of controlled randomized human trials on vitamin E inconsistent and disappointing?
EVIDENCE FOR LIPID PEROXIDATION IN VIVO AND ITS ASSOCIATION WITH DISEASE
Polyunsaturated fatty acids such as linoleic acid and arachidonic acid and their esters are highly susceptible to oxidation. They are oxidized into diverse products, some of which are cytotoxic and sufficiently reactive to modify proteins and DNA bases [9]. Lipids are oxidized by enzymes, free radicals, and nonenzymatic, nonradical oxidants. Enzymatic oxidation is often induced in a regulated manner by lipoxygenase, cyclooxygenase, and cytochrome P450 to yield specific physiologically essential products. On the other hand, oxidation mediated by free radicals, which is termed lipid peroxidation, proceeds randomly and nonspecifically.
Free radicals attack proteins and DNA, as well as lipids, nonselectively. Therefore, when free radicals are formed in vivo by, for example, high-energy irradiation or ischemia-reperfusion injury, the levels of oxidation products of proteins and DNA, as well as those of lipid peroxidation, are increased. Furthermore, secondary lipid peroxidation products, such as unsaturated aldehydes, readily react with protein thiols, resulting in loss of protein function and cellular homeostasis. Among the lipid peroxidation products, hydroxyoctadecadienoic acid (HODE) from linoleic acid, and hydroxyeicosatetraenoic acid (HETE) and isoprostanes from arachidonic acid, have frequently been used as in vivo biomarkers of lipid peroxidation [10].
As discussed later, vitamin E is a potent radical-scavenging antioxidant that inhibits lipid peroxidation mediated by free radicals but not enzymatic oxidation by lipoxygenase and cyclooxygenase. HODE and HETE are produced by enzymatic oxidation as well as free radical oxidation, but their isomer distribution depends on the type of oxidant. Therefore, it is important to understand the mechanisms and oxidants of lipid oxidation to evaluate the effects of vitamin E. The mechanisms of lipid oxidation have been investigated extensively and are now well understood [9,11]. It has been shown that trans, trans-forms of HODE and HETE are specific products of lipid peroxidation mediated by free radicals.
Numerous studies have shown that in general, if not always, the levels of lipid peroxidation products, such as HODE, HETE, and isoprostanes, in biological fluids and tissues of diseased patients are higher than in healthy subjects. Furthermore, clinical studies have established a link between disease states and lipid peroxidation products.
Two examples are described below.
Liver diseases
The importance of lipid peroxidation mediated by free radicals in liver injury induced by carbon tetrachloride and other halogenated alkanes has been investigated since the 1960s and documented in detail [12]. It was once thought that carbon tetrachloride affects the liver by the action of a simple solvent, but it is now understood that carbon tetrachloride must undergo metabolic activation to trichloromethyl radical by cytochrome P450, primarily by cytochrome P450 2E1, to exert its toxic effect [13]. The trichloromethyl radical reacts rapidly with oxygen to yield the trichloromethyl peroxyl radical, which attacks lipids and induces their peroxidation.
Lipid peroxidation mediated by free radicals is involved in alcoholic liver disease caused by chronic alcohol ingestion [14,15]. Plasma levels of several lipid oxidation products, including oxysterols and isoprostanes, have been shown to be elevated in alcoholic liver disease patients [16,17].
Nonalcoholic fatty liver disease (NAFLD), a hepatic manifestation of metabolic syndrome, is now the most common liver disorder, affecting a high proportion of the global population. The incidence of NAFLD is increasing due to increases in the prevalence of two major risk factors, obesity, and type 2 diabetes, which are related to lifestyle and diet. The characteristic feature of NAFLD is an excessive accumulation of fat, notably triglyceride, in the liver, and it encompasses a wide spectrum from benign steatosis to nonalcoholic steatohepatitis (NASH), liver cirrhosis, liver failure, and hepatocellular carcinoma [18].
NAFLD and NASH are multifactorial diseases and oxidative stress has been implicated in their pathogenesis. Several human and animal studies have reported an association between NAFLD/NASH disease state and biomarkers of lipid peroxidation [19]. One such study reported that levels of 9- and 13-HODE, major products of linoleic acid peroxidation, were significantly elevated in patients with NASH compared to those with steatosis, and a strong correlation was observed between these oxidation products and liver histopathology such as inflammation, fibrosis, and steatosis [20]. These HODEs were racemic, suggesting them to be produced by free radical oxidation.
Interestingly, intraperitoneal administration of 2,2’-azobis(2-amidinopropane) dihydrochloride (AAPH), an azo compound that produces free radicals spontaneously by thermal decomposition without biotransformation, induces fatty liver in mice [21,22]. The administration of AAPH to mice and rats increases levels of lipid peroxidation products such as HODE, HETE, and isoprostanes [22,23]. Collectively, the experimental evidence shows the involvement of radical-mediated lipid peroxidation in liver diseases.
Numerous studies have reported a beneficial effect of vitamin E on NAFLD and NASH [19]. In one such study, the effects of vitamin E at a dose of 800 IU/day or placebo for 96 weeks were examined in adults with NASH and without diabetes; there was improvement in the histological features of NASH [24]. Another study reported measurable differences in the metabolic profile of subjects likely to respond to vitamin E treatment for NASH and those who experienced histological improvements following treatment [25]. In a recent retrospective study of the effects of 300 mg/day vitamin E for 2 or more years in patients with biopsy-proven NASH, vitamin E ameliorated NASH fibrosis, especially in those who showed improved transaminase activities and insulin resistance [26].
Atherosclerosis
Atherosclerosis is a major cause of stroke, myocardial infarction, coronary artery disease, and peripheral vascular disease. It is a slowly progressive and cumulative inflammatory disease characterized by excessive deposition of cholesterol in the arterial wall. It begins in childhood and remains asymptomatic for decades, but is the leading cause of death in industrialized countries.
It is generally accepted that oxidative modification of low density lipoprotein (LDL) is an important initial event in atherosclerosis [27]. Incubation of macrophages with oxidized LDL, but not native LDL, leads to the intracellular accumulation of cholesteryl esters. The oxidation of LDL increases its pro-atherogenic effect, whereas that of high density lipoprotein diminishes its anti-atherogenic effect [28].
The oxidative modification of LDL was investigated extensively in the 1990s and its mechanisms and products have been elucidated, although the epitope responsible for recognition by macrophage scavenger receptors has not been fully characterized. Nevertheless, it has been shown that oxidatively modified LDL is taken up by macrophages, which is the initial step in the formation of foam cells.
Oxidation of LDL yields diverse products. Cholesteryl esters and phosphatidylcholine (PC) are the major lipids in human LDL particles. Linoleic acid and arachidonic acid esters of cholesterol and PC are oxidized into the corresponding hydroperoxides, hydroxides, and breakdown products [29]. The levels of lipid peroxidation products in the plasma of atherosclerotic patients are higher than those of healthy subjects. HODE and HETE levels are higher in LDL isolated from diabetic patients than that from healthy subjects. The molecular ratios of HODE and HETE to the parent lipids (linoleates and arachidonates, respectively) are also higher in diabetic LDL than in control LDL [30]. Moreover, the levels of oxysterols, such as 7-b-hydroxycholesterol, 7-ketocholesterol, and cholesterol-5,6-epoxide, increase in the order normal artery < fatty streak < advanced lesion [31]. Furthermore, cholesteryl linoleate is often present in hydroperoxide and hydroxide forms in human atherosclerotic plaque [32,33].
It should be pointed out that the oxidative modification of LDL is mediated by several oxidants and by different mechanisms. LDL is oxidized by free radicals and also by nonradical oxidants such as lipoxygenases [34], cytochrome P450 enzymes, and hypochlorite [35]. Singlet oxygen may also contribute [36]. Importantly, different oxidants produce characteristic and specific oxidation products, and thus require different antioxidants. No single antioxidant can inhibit all types of oxidation.
ANTIOXIDANT ACTION OF VITAMIN E
Vitamin E is the most abundant and potent radical-scavenging antioxidant in vivo. It is lipophilic and had antioxidant effects within biological membranes and lipoproteins in concert with the hydrophilic vitamin C. Vitamins E and C inhibit oxidation of LDL in a synergistic manner [37,38]. The mechanisms and dynamics of action of vitamin E as a radical-scavenging antioxidant have been investigated extensively and are well documented [39,40].
The unequivocal biological action of vitamin E as an antioxidant was first demonstrated against carbon tetrachloride toxicity in liver damage [41]. As mentioned above, carbon tetrachloride induces lipid peroxidation mediated by free radicals and leads to liver damage, which is inhibited by prior administration of vitamin E. Numerous studies have confirmed the antioxidant effects of vitamin E and other antioxidants against carbon tetrachloride toxicity in cell culture and animal experiments.
When vitamin E scavenges peroxyl radicals, it is converted into vitamin E radical, which may be further oxidized into α-tocopheryl quinone or reduced by vitamin C or other reducing compounds to regenerate vitamin E. α-Tocopheryl quinone is a biomarker of the antioxidant action of vitamin E [42]. Interestingly, a high level of α-tocopheryl quinone is found in human atherosclerotic plaque [32].
Vitamin E deficiency may lead to various disorders such as ataxia, neurological disorders, and infertility. Some disorders can be rescued by synthetic radical-scavenging antioxidants [43], suggesting a role for vitamin E as a radical-scavenging antioxidant.
Limitations of vitamin E
As mentioned above, oxidation of biological molecules is mediated by multiple oxidants. Vitamin E is a potent radical-scavenging antioxidant, but it is ineffective against nonradical oxidants such as lipoxygenase, cyclooxygenase, cytochrome P450, and hypochlorite [44]. These oxidants may be involved in the pathogenesis of diseases such as atherosclerosis, and their relative contributions may vary depending on the circumstances. This suggests that the effects of vitamin E against oxidative stress-induced diseases are limited and that even large-scale, randomized clinical trials of vitamin E or any other single antioxidant may not provide clear results. Multiple antioxidants that are active against different oxidants are required to cope with oxidative stress in vivo.
EVIDENCE FOR BENEFICIAL EFFECTS OF VITAMIN E ON DISEASES
Lipid peroxidation mediated by free radicals has been implicated in the pathogenesis of various diseases. This implies that vitamin E and other antioxidants that inhibit lipid peroxidation may reduce the disease risk or be useful in disease prevention and treatment. Indeed, vitamin E has been found to have beneficial effects in animal models of various diseases [40]. In contrast, the effects of vitamin E in humans are less consistent and have often been disappointing.
The effects of vitamin E supplementation on biomarkers of oxidative stress in humans have been investigated. Antioxidant concentrations in human plasma are increased by supplementation, and the levels of oxidative stress biomarkers are decreased, especially for those experiencing oxidative stress; however, the effects on markers of systemic inflammation are limited [45]. At the same time, the level of oxidative stress in well-nourished, healthy subjects doe not change significantly [46].
Epidemiologic studies have reported that individuals who consume high amounts of vitamin E through diet or supplements have decreased rates of chronic diseases such as cardiovascular disease [47,48]. However, many large, prospective, randomized, placebo-controlled clinical trials of vitamin E have yielded disappointing and conflicting results. Furthermore, some meta-analyses have suggested that high-dose vitamin E actually increases all-cause mortality [49].
Oxidative modification of LDL is an important initial event in atherogenesis. Consequently, vitamin E is expected to have beneficial effects, but many randomized clinical trials of vitamin E have yielded disappointing results in terms of both mortality and morbidity. It is now thought that indiscriminate supplementation with high doses of vitamin E is not beneficial for preventing cardiovascular disease [50]. More recently, the Korean Meta Analysis Study Group reported that there is no evidence to support the use of vitamin E and antioxidant supplements for the prevention of cardiovascular diseases [51].
Possible reasons for failure of vitamin E clinical trials
There are several reasons for the disappointing and inconsistent results of vitamin E treatment.
First, an oxidative event may a consequence, rather than a cause, of disease or a concomitantly occurring phenomenon, with no cause-consequence relationship. Many diseases are not caused by oxidative stress alone. However, the results of abundant in vitro and animal experiments and the close association between the levels of oxidative stress and the disease state reported in many studies strongly support the notion that free radical-mediated oxidation of biological molecules plays a causative role in the pathogenesis of some diseases.
Second, multiple oxidants with different reactivities and selectivities may contribute to disease etiology. No single antioxidant can cope with all of them, and thus multiple antioxidants with different functions and selectivity are required. As mentioned above, vitamin E is a potent antioxidant against free-radical oxidation but not against enzymatic oxidation, which may also contribute to disease pathogenesis. The relative contributions of free-radical and non-free-radical oxidants, and hence the efficacy of vitamin E, may vary.
Third, the subjects included in previous studies may not have been appropriate. For example, healthy subjects with adequate antioxidant levels may limit the potential to benefit from supplemental antioxidants. Finally, many studies did not assess the level of oxidative stress in subjects.
The importance of proper subject selection was demonstrated by Vardi and colleagues [52]. They found that patients with diabetes mellitus and haptoglobin (Hp) genotype 2-2 are under more severe oxidative stress than those with Hp genotype 1-2 or 1-2. Two classes of alleles exist at the Hp locus, denoted 1 and 2, for three possible Hp genotypes: 1-1, 2-1, and 2-2. Hp stabilizes hemoglobin, Hp 2 being less potent in this regard than Hp 1. Another study found that vitamin E was beneficial to Hp 2-2 diabetic individuals [53]. A polymorphism in the haptoglobin gene may determine the efficacy of vitamin E treatment.
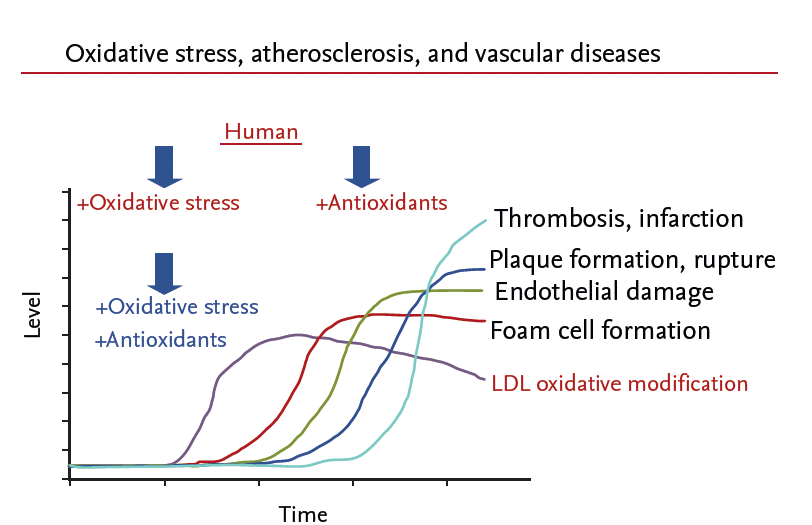
Timing is also an important issue. Oxidative stress may be pivotal for the initiation of diseases but becomes progressively less important during the later stages of chronic diseases, including atherosclerosis and neurodegenerative conditions. The subjects in human trials are in general older than 50 years, which may be too old for antioxidant treatment to have a beneficial effect. On the other hand, in many animal studies, vitamin E is given simultaneously with the onset of oxidative stress (Fig. 1), which may explain the greater beneficial effect in animals than humans.
The dosage and duration of vitamin E treatment are also important factors. These are often determined arbitrarily and the optimum dosage is difficult to determine. It has been pointed out that concurrent use of vitamin C may be necessary.
The lack of biomarkers for oxidative stress and the monitoring of vitamin E levels has also been pointed out. Poor subject compliance may be another issue.
Oxidative stress may not always be a detrimental phenomenon. It is now accepted that ROS and oxidative stress have important roles in physiology as well as pathophysiology. Some ROS are produced in a regulated manner to play a role as physiological signaling messengers that maintain homeostasis [54]. A low level of oxidative stress is a positive factor, known as eustress.
Furthermore, recent studies have shown that lipid peroxidation products are also capable of acting as signaling mediators and inducers of an adaptive response to upregulate defense capacity [9], in many cases, through the nuclear factor erythroid 2 related factor 2-Kelch-like ECH-associated protein 1 system [55]. Recently, it has been argued that excess antioxidants may remove too many ROS and impair ROS/reactive nitrogen species (RNS) signaling and lipid peroxidation, and that antioxidants at high doses may be harmful [56]. However, it should be emphasized that the radical-scavenging antioxidants (such as vitamin E and vitamin C) do not scavenge physiologically important signaling ROS, such as superoxide and hydrogen peroxide, nor do they inhibit enzymatic lipid oxidation [57]. These antioxidants are not potent inhibitors of myeloperoxidase-mediated reactions [58]. It is unlikely that these antioxidants impair physiological signaling by ROS/RNS, even at high doses.
For ROS to function as physiologically essential signaling molecules, their formation, reaction, and metabolism must be strictly regulated and controlled. The lack of regulation and specificity of free radical formation and reactions make it difficult for lipid peroxidation products to act as physiologically essential signaling molecules. The formation of lipid peroxidation products cannot be programmed or regulated. The adaptive response induced by lipid peroxidation products should be considered a protective response of the organism to xenobiotics. Inhibition of radical-mediated lipid peroxidation by vitamin E should have a beneficial effect.
Does vitamin E have adverse effects?
Some studies have reported adverse effects of large doses of vitamin E supplements. As mentioned above, a meta-analysis of 19 clinical trials with 135,968 participants showed that high-dosage vitamin E supplements (≥ 400 IU) might increase all-cause mortality [49]. On the other hand, a recent review article reported that a pooled analysis of 18 randomized controlled trials undertaken in apparently healthy individuals showed no effects of vitamin E supplementation at a dose of 23 to 800 IU/day on all-cause mortality [59]. Moreover, in meta-analyses of 33 and 57 trials, vitamin E supplementation was not correlated with mortality [60,61].
The mechanism underlying the increased risk of high-dose vitamin E supplementation is unknown. The phenomenon may be due to the induction of cytochrome P450, which accelerates metabolism of other drugs [62]. Vitamin E has eight isoforms, α-tocopherol being the most potent and abundant in humans, although a similar quantity of α-tocopherol is consumed in the diet. Supplementation with high-dose α-tocopherol accelerates the metabolism of non-α-tocopherol forms. It has not been demonstrated whether or not the various isoforms of vitamin E have specific functions in vivo, which might be impaired by α-tocopherol supplementation.
In certain in vitro systems, it has been found that α-tocopherol may act as a pro-oxidant. For example, α-tocopherol accelerates oxidation of LDL in the absence of reducing agents such as vitamin C, because the a-tocopheroxyl radical induces LDL oxidation. However, vitamin C completely inhibits this pro-oxidant action by reducing levels of the α-tocopheroxyl radical [63]. Therefore, it is unlikely that vitamin E acts as a pro-oxidant in vivo.
CONCLUSIONS
There is ample evidence that lipid peroxidation mediated by free radicals is involved in the pathogenesis of various diseases, that vitamin E is a potent radical-scavenging antioxidant, and that vitamin E inhibits lipid peroxidation in vivo as well as in vitro. These findings suggest that vitamin E should reduce the risk, or be useful in the prevention and treatment, of diseases mediated by free radicals. Many epidemiological studies have supported this notion, but the results of clinical intervention studies and meta-analyses have been controversial; some have reported positive findings, many have reported null findings, and some have yielded adverse findings.
Various factors must be considered if optimal results are to be achieved. Vitamin E should be beneficial to subjects experiencing oxidative stress mediated by free radicals when given in the correct dosage, at the right time, and for the appropriate duration.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





