Guidelines for the management of myeloproliferative neoplasms
Article information
Abstract
Polycythemia vera, essential thrombocythemia, and primary myelofibrosis are collectively known as ‘Philadelphia-negative classical myeloproliferative neoplasms (MPNs).’ The discovery of new genetic aberrations such as Janus kinase 2 (JAK2) have enhanced our understanding of the pathophysiology of MPNs. Currently, the JAK2 mutation is not only a standard criterion for diagnosis but is also a new target for drug development. The JAK1/2 inhibitor, ruxolitinib, was the first JAK inhibitor approved for patients with intermediate- to high-risk myelofibrosis and its effects in improving symptoms and survival benefits were demonstrated by randomized controlled trials. In 2011, the Korean Society of Hematology MPN Working Party devised diagnostic and therapeutic guidelines for Korean MPN patients. Subsequently, other genetic mutations have been discovered and many kinds of new drugs are now under clinical investigation. In view of recent developments, we have revised the guidelines for the diagnosis and management of MPN based on published evidence and the experiences of the expert panel. Here we describe the epidemiology, new genetic mutations, and novel therapeutic options as well as diagnostic criteria and standard treatment strategies for MPN patients in Korea.
INTRODUCTION
Chronic myelogenous leukemia, polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) are myeloproliferative disorders characterized by clonal expansion of abnormal hematopoietic stem/progenitor cells. The term ‘myeloproliferative disorders’ was replaced by ‘chronic myeloproliferative diseases’ in 2001, and now ‘myeloproliferative neoplasms’ (MPNs) is standard terminology according to 2008 World Health Organization (WHO) criteria [1]. Among these four disease entities, PV, ET, and PMF are collectively called ‘Philadelphia-negative classical MPNs.’ The discovery of new genetic aberrations including Janus kinase 2 (JAK2) and thrombopoietin receptor (MPL) enhanced our understanding of MPN pathophysiology. Currently, the JAK2 mutation is not only a standard criterion for diagnosis but also a new target for drug development.
According to data from the Health Insurance Review and Assessment Service (HIRA) of Korea, there was a 3.8-fold increase in the number of registered MPN cases over a 10-year period. Considering the prolonged lifespan of Koreans, the prevalence of MPNs would be expected to increase gradually year by year. However, established therapeutic options have been quite limited to date. Many kinds of new drugs for treating MPNs are under clinical investigation, but these new drugs have not yet been incorporated into standard management. Hence, treatment of Philadelphia-negative classical MPNs remains challenging [2].
Previously, we summarized diagnostic and therapeutic guidelines for Korean MPN patients [3]. In view of recent developments, we have revised the guidelines for the diagnosis and management of MPN based on published studies and the experiences of the expert panel.
DIAGNOSIS OF MPN
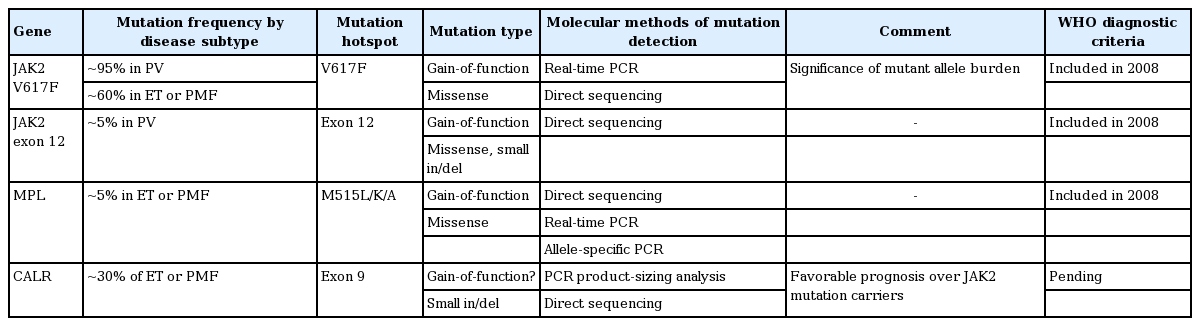
Diagnosis of MPN according to the revised 2008 WHO criteria is based on a combination of clinical, morphological, and molecular parameters (Table 1) [4]. Cardinal features of Philadelphia chromosome-negative MPN are increased red blood cell (RBC) production in PV, sustained thrombocytosis in ET, and bone marrow (BM) fibrosis in PMF. Molecular markers, such as JAK2 V617F, JAK2 exon 12, and MPL mutations, have increased our knowledge of the pathogenesis of MPNs as well as the accuracy of diagnosis. Furthermore, many recent studies have reported that the calreticulin mutation (mutation in exon 9 of CALR) is found in the majority of patients with MPN with nonmutated JAK2 [5,6]. There are many controversies with regard to the subjectivity and the lack of reproducibility of the histological criteria. And BM assessment is not always needed, especially in PV. However, histologic evaluation of the pathologic features of megakaryocytic, granulocytic, and erythroid series and cellularity is important in differentiating MPNs, especially ET and PMF [7]. Sometimes, patients with occult MPN present with normal blood counts because of gastrointestinal bleeding and associated iron deficiency, splenomegaly, or simply because they are in an early stage of the disorder [8]. In cases of clinically suspicious MPN, such as intra-abdominal thrombosis with normal blood counts, careful investigation is needed [9].

World Health Organization 2008 diagnostic criteria for myeloproliferative neoplasm
Polycythemia vera
PV is characterized by increased RBC production independent of the mechanisms that regulate erythropoiesis [4]. Characteristic features are increased hemoglobin, hypercellular BM with panmyelosis, decreased serum erythropoietin, and the presence of the JAK2 mutation. A BM study is not needed for diagnosis of PV if two of the major criteria are fulfilled [10]. However, BM biopsy is recommended because the degree of fibrosis confers valuable prognostic information [11].
Primary myelofibrosis
PMF is characterized by a proliferation of predominantly megakaryocytes and granulocytes in the BM that in fully developed disease is associated with reactive deposition of connective tissue and extramedullary hematopoiesis [4]. Characteristic features are megakaryocytic proliferation with atypia and fibrosis or hypercellular marrow with megakaryocytic and granulocytic proliferation; characteristic blood features such as leukoerythroblastosis, tear drop cells, and anemia; splenomegaly; increased levels of lactate dehydrogenase (LDH); and JAK2 mutation. In the early phase (prefibrotic stage), the only finding may be marked thrombocytosis so that differential diagnosis with ET is not always clear [12].
Essential thrombocythemia
ET usually involves primarily the megakaryocytic lineage. Characteristic features are sustained thrombocytosis (> 450 × 109 /L), megakaryocytic proliferation, and JAK2 mutation [4]. When the WHO classification is applied to thrombocytosis patients, prefibrotic PMF should be excluded before diagnosis of ET. Diagnosis of ET and PMF in patients lacking a molecular marker is challenging, but a CALR (calreticulin) gene mutation has been detected that is almost always seen in JAK2 V617F-negative and MPL-non-mutated patients. Therefore, some investigators have proposed that CALR should be added to the criteria for ET and PMF diagnosis [13].
MOLECULAR GENETICS OF MPNs
JAK2 mutations
JAK2 V617F is observed across a broad spectrum of MPNs including PV, ET, and PMF [1,14]. The JAK2 protein has tyrosine kinase activity and the gain-of-function V617F mutation results in constitutive activation of the JAK/STAT pathway. The burden of the mutant allele has been reported to be associated with disease subtype and prognosis [15,16]. In PV patients without JAK2 V617F (~5%), a somatic mutation is detected in exon 12 of JAK2 [17,18].
MPL W515L/K/A mutations
Approximately 5% of patients with ET or PMF have a somatic MPL W515L/K/A mutation [1]. MPL encodes the receptor for thrombopoietin, the major regulator of platelet production. W515L/K/A mutations are gain-offunction mutations inducing constitutive signaling of the downstream JAK/STAT pathway. Korean data have shown that ~7% of ET patients without JAK2 mutations have W515L/K mutations [19]. No clinical relevance has been reported regarding the mutant burden.
CALR mutations
Mutations in the CALR gene have recently been reported in a significant proportion of MPN patients (~70% of JAK2-nonmutated ET and ~85% of JAK2-nonmutated PMF) [5,6,20]. Calreticulin is a protein with multiple functions including roles in cell proliferation and apoptosis. Mutations occurring in exon 9 are most commonly small deletions or insertions with or without substitutions. The mutations result in a frameshift to the alternative reading frame, and the resultant mutant proteins have a novel amino acid sequence at the C-terminus. Pathologically, CALR mutations are confined to ET, PMF, and refractory anemia with ringed sideroblasts associated with marked thrombocytosis (not seen in PV). Clinically, patients with CALR mutations were reported to have higher platelet counts, lower leukocyte and hemoglobin levels, lower risk of thrombosis, and a more indolent clinical course compared to those with JAK2 mutations [21]. The significance of the CALR mutant allele burden is not known. CALR mutations are expected to be assimilated into the diagnostic criteria for MPNs (especially ET and PMF) in the upcoming WHO classification.
Other mutations
Other genes reported to be recurrently mutated in MPNs include those involved in epigenetics (TET2, DNMT3A, ASXL1, IDH1/2, and EZH2) and splicing (SF3B1 and SRSF2) and oncogenes (TP53, NRAS, and KRAS) [6,22]. The clinical utility of mutation detection in these genes is currently limited due to: (1) the low frequency; (2) poor genotype-phenotype correlations; and (3) uncertain clinical significance. Lastly, it is important to rule out a hereditary background [23]. Recent data in Korea revealed a significant proportion (13.2%) of patients with JAK2-negative idiopathic isolated erythrocytosis had germline mutations [24].
Proposal for molecular genetics testing
JAK2, CALR, or MPL gene mutations are detected in > 90% of patients with non-BCR/ABL1 classical MPN, notably in a mutually exclusively way (Table 2). Thus, the first diagnostic molecular test is a quantitative test for JAK2 V617F. When JAK2 V617F is negative and PV is suspected, the second-line test is for the JAK2 exon 12 mutation by direct sequencing. When JAK2 V617F is negative and ET or PMF is suspected, the second-line targets are CALR and MPL mutations. Given the genetic information (mutated gene, type of mutation, and mutation burden), genotype-phenotype correlations are needed in terms of laboratory parameters, BM pathology, and clinical course. Molecular genetic tests to search for a hereditary background are needed in JAK2-negative isolated erythrocytosis.
Summary of major gene mutations in the diagnostic workup of BCR/ABL1-negative classical myeloproliferative neoplasms
EPIDEMIOLOGY
Domestic epidemiologic data for MPN can be estimated from the number of cases registered at the HIRA of Korea, which are gathered as claims for reimbursements of test fees and drug prescriptions for each diagnostic category. The HIRA is a government-affiliated organization that constructs an accurate claims review and quality assessment system for the National Health Insurance (NHI). The NHI program covers the entire Korean population as a compulsory social insurance system. Age-standardized incidence rates are estimated by calculating weighted averages of crude age-specific rates, using the mid-year estimated population of Korea in the year 2000 as the standard population, and calculated using the world standard population as the reference population [24].
The most notable result is that the number of MPN cases registered to the HIRA showed a 3.8-fold increase during a 10-year period (Fig. 1). The probable attributable causes for the increase include: more accurate diagnosis of MPN with the introduction of detection methods for the JAK2 mutation; the increase in average lifespan over the years; and the introduction of the registration system for rare or intractable diseases for central insurance coverage. However, the CALR mutation [5,6] was recently discovered in JAK2-negative myelofibrosis (MF) and ET patients, which may lead to a further increase in the number of MPN diagnoses in the future.

Annual registered number of patients with polycythemia vera, essential thrombocythemia and myelofibrosis from the databases of the Korean Health Insurance Review and Assessment Service.
By age, PV, MF, and ET were all most prevalent in the seventh decade of life, but in the most recent 2 to 3 years (years 2009 to 2011), MF and ET were most prevalent in the eighth decade in females. The male-to-female ratios for PV, MF, and ET were 1.5, 1.0, and 0.7, respectively, which are comparable to those documented in Western countries. Incidence rates in the year 2007, standardized according to the mid-year population in the year 2000, for PV, ET, and MF were 2.9, 2.2, and 1.4 per 100,000 persons, respectively. However, the actual incidence rates observed by clinicians in the field are higher for ET than PV. The reason for this distortion seems to have been the false inclusion of secondary erythrocytosis under the PV diagnosis category in the years before JAK2 mutation testing was largely available. This inference is proven by the fact that the incidence rate of ET (5.3/100,000) was higher than PV (3.3/100,000) in 2011.
PRIMARY MYELOFIBROSIS
Clinical features
The clinical features of PMF and secondary MF are similar; hence, these two conditions are indistinguishable by symptoms or signs. Regardless of whether the diagnosis is primary or secondary, clonal proliferation in the BM is associated with leukoerythroblastosis in peripheral blood smears and extramedullary hematopoiesis, which causes hepatosplenomegaly. Laboratory findings include progressive anemia, leukocytosis or leukopenia, thrombocytosis or thrombocytopenia, and increased levels of LDH. Inflammatory and angiogenic cytokines, such as transforming growth factor β (TGF-β), vascular endothelial growth factor, interleukin 1 (IL-1), IL-2, and IL-6, are increased in MF patients, and this is related to the pathogenesis of portal hypertension or pulmonary hypertension in MF [25-28]. Constitutional symptoms (fever, night sweats, weight loss) are more frequently reported in MF patients compared to those with PV or ET [29]. The Myeloproliferative Neoplasm Symptom Assessment Form total symptom score (MPN-SAF TSS) is a simple assessment tool for checking the patient’s constitutional symptoms at diagnosis and during follow-up [30]. Aggravation of splenomegaly (pain, early satiety, symptoms of portal hypertension) and constitutional symptoms are shown with progression of MF.
Prognosis and risk stratification
The median survival of patients diagnosed with MF ranges from 4 to 5.5 years. The main causes of death are progression to acute leukemia, infection/hemorrhage due to BM failure, and complications related to portal hypertension. Selection of treatment depends on the risk to the individual patient. Prognostic scoring was initiated with the International Prognostic Scoring System (IPSS) in 2009. It includes five clinical risk factors checked at diagnosis (age > 65 years, hemoglobin < 10 g/dL, white blood cell [WBC] count > 25 × 109/L, circulating blasts > 1%, presence of constitutional symptoms) and provides four categories of risk (a score of 0 for low risk, 1 for intermediate risk-1, 2 for intermediate risk-2, and 3 or more for high risk). Overall survival for each risk group is 135, 95, 48, and 27 months, respectively [31]. The Dynamic International Prognostic Scoring System (DIPSS) uses the same values as IPSS but assigns a higher score for anemia. Risk categorization is also modified from that of the IPSS (score 0 for low, 1 or 2 for intermediate-1, 3 or 4 for intermediate-2, 5 or 6 for high). Median survival for each corresponding risk group of DIPSS is ‘not reached,’ 14.2, 4.0, and 1.5 years, respectively. Scoring via DIPSS can occur at any time during the patient’s progress [32]. Gangat et al. [33] has refined DIPSS to DIPSS-plus with the inclusion of three additional risk factors: thrombocytopenia < 100 × 109/L, RBC transfusion requirement, and unfavorable karyotypes (complex karyotype or sole or two abnormalities including +8, -7/7q-, i(17q), inv(3), -5/5q-, 12p-, 11q23 rearrangement). The median overall survival of each DIPSS-plus risk group is 15.4, 6.5, 2.9, and 1.3 years, respectively [33]. These three risk stratifications are all currently used for choosing risk-adapted treatment for MF patients.
Genetic mutations have prognostic values independent of DIPSS-plus. A mutation of at least one of the five genes (ASXL1, EZH2, SRSF2, IDH1, and IDH2) is defined as a “high-molecular risk (HMR) category.” HMR is related to the risk of death and leukemic transformation and an increased HMR ‘number’ also correlates with leukemic progression [34,35]. However, JAK2, MPL, and TET2 mutations were shown to have little influence on prognosis [30,36,37]. Elevated circulating cytokines (IL-8, IL-2R) are also correlated with an unfavorable prognosis regardless of the DIPSS-plus score [38].
Response criteria
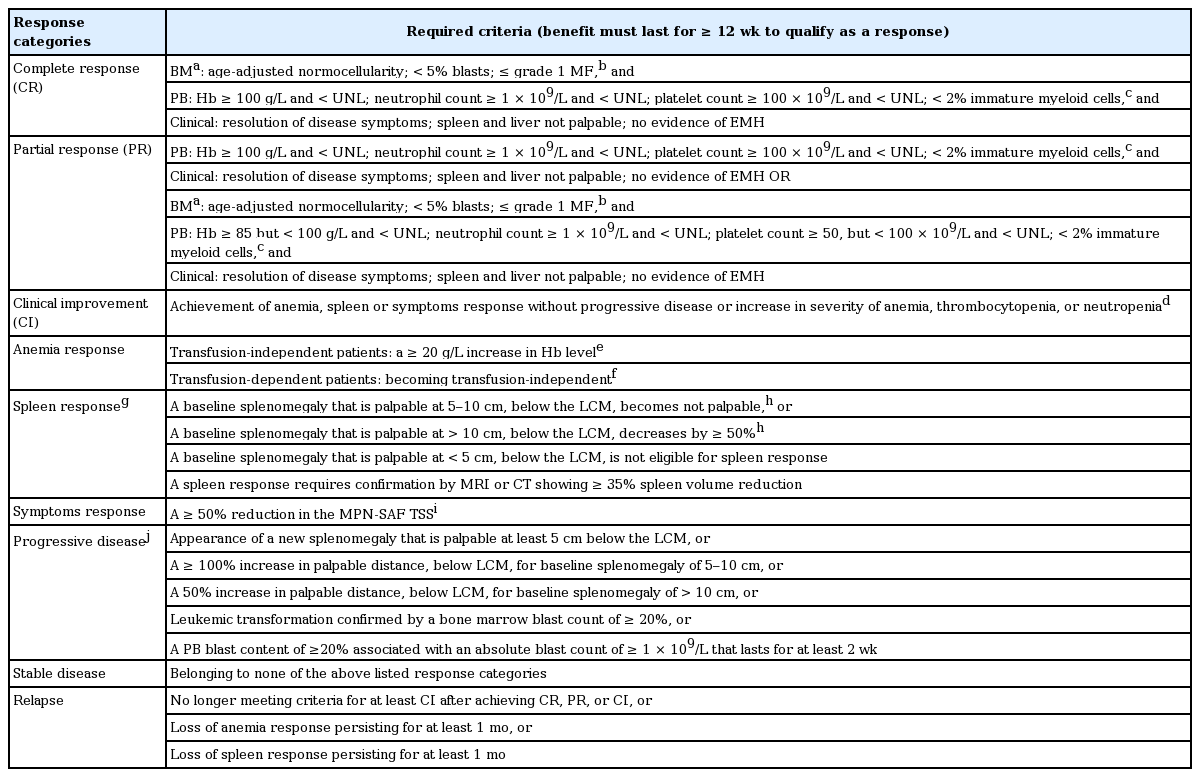
Recently, the International Working Group-Myeloproliferative Neoplasms Research Treatment (IWG-MRT) and European LeukemiaNet (IWG-MRT-ELN) revised their response criteria for MF patients [39].
The 2013 revision included a strict definition of RBC transfusion dependency and introduced the MPN-SAF TSS [40] as a quantifiable method for scoring disease-related symptoms. MPN-SAF TSS [40] is calculated as the mean score for 10 items (fatigue, concentration, early satiety, inactivity, night sweats, itching, bone pain, abdominal discomfort, weight loss, and fever) adapted from the Brief Fatigue Inventory [41] and MPN-SAF self-assessment [42]. Each item is scored on a 0 (absent/as good as it can be) to 10 (worst imaginable/as bad as it can be) scale, and MPN-SAF TSS is computed as the average of the observed items multiplied by 10 to achieve a 0-to-100 total scale [40]. Since several clinical trials demonstrated that anemia [43] and spleen [31,44] responses are well correlated with patients’ quality of life, they suggested anemia and spleen responses be included in the additional composite response category along with the assessment of MPN-SAF TSS [39]. Moreover, they recommended new response criteria to assess cytogenetic and molecular remissions with treatment [39].
Response criteria in this guideline are derived from the 2013 IWG-MRT-ELN response assessment with minimal modifications (Table 3) [45]. The 2013 IWG-MRT-ELN guidelines recommended molecular monitoring for MF patients [39] every 6 months. On the other hand, the Nordic MPN group did not recommend routine BM examination or routine monitoring of molecular responses during treatment of MF patients without a stem cell transplant setting [46]. The role of reduction of the JAK2 V617F allele burden in clinical findings or treatment outcomes of MF patients remains to be elucidated [36,47,48]. However, a recent clinical trial demonstrated that JAK2 V617F allele burden is a predictive factor of treatment outcome and risk of relapse in stem cell transplanted patients with MPNs [49]. Hence, routine molecular monitoring in PMF patients can be reserved for experimental or stem cell transplant settings.
Response criteria for myelofibrosis
Traditional treatment approaches
Stem cell transplantation
Allogeneic stem cell transplantation (allo-SCT) is the only curative treatment in PMF. However, transplant-related mortality and severe morbidity occur in half of patients. In a large study of allo-SCT in PMF, 5-year disease-free survival and treatment-related mortality were 33% and 35% for matched related and 27% and 50% for unrelated transplants, respectively [50]. Due to its significant toxicity, allo-SCT should be considered in patients with an expected survival of less than 5 years. Therefore, allo-SCT is recommended in patients assessed as intermediate-2 and high risk at diagnosis, and during follow-up of younger low and intermediate-1 patients who progress to a higher risk by DIPSS or DIPSS plus evaluation [2,51-54]. In terms of a conditioning regimen, patients over 40 or 45 years have exhibited very poor survival after myeloablative conditioning, but reduced-intensity conditioning has improved results in those patients [55]. Therefore, reduced-intensity transplantation should be considered for patients aged 40 or over.
Autologous stem cell transplantation (auto-SCT) demonstrated only a modest benefit and little therapeutic efficacy in a previous study; hence, auto-SCT is not recommended in PMF patients [56].
Treatment of anemia
Erythropoiesis-stimulating agents (ESAs) can benefit PMF patients. ESAs have been shown to effectively increase hemoglobin levels in 20% to 60% of PMF patients. A plasma erythropoietin level below 125 U/L has been associated with a higher probability of response [57]. Corticosteroids such as prednisone may be temporarily effective for treatment of symptoms [54,58]. Androgenic steroids such as danazol may stimulate BM function and have been shown to improve hemoglobin concentration in 40% of patients [59]. Thalidomide [60] or lenalidomide [61] in combination with low-dose prednisone can increase hemoglobin levels and decrease spleen size. However, drug adverse effects—including hepatotoxicity and virilizing effects for androgens, peripheral neuropathy for thalidomide, and myelosuppression for lenalidomide—should be monitored [54,62].
Treatment of splenomegaly and constitutional symptoms
Hydroxyurea is one of the most commonly used agents in MPN patients. According to previous studies, hydroxyurea is an effective and generally well-tolerated therapy for hyperproliferative manifestations of MF [63]. Hydroxyurea resulted in improvement in splenomegaly, bone pain, constitutional symptoms, and pruritus [63,64]. However, the improvement in symptoms with hydroxyurea is temporary and adverse effects of treatment include myelosuppression and mucocutaneous ulcers [54,62]. Anagrelide may be used in PMF patients with symptomatic thrombocytosis, but this agent does not inhibit progression of MF [46,53]. Interferon α (IFN-α) may be efficacious in PMF, especially in patients with the hyperproliferative stage of the disease, and pegylated-IFN-α2a has been investigated in the treatment of MF [65,66]. In contrast, patients with advanced disease showed a lower response rate to this agent. Moreover, pegylated-IFN-α2a appears to have little clinical effect in reducing splenomegaly, but has a role as a myelosuppressive agent.
Given the high complication rate and limited benefit of splenectomy, patient selection is crucial. Splenectomy should be restricted to carefully selected patients with refractory hemolysis, drug-refractory symptomatic splenomegaly, significant splenic infarction, symptomatic portal hypertension (e.g., ascites, bleeding varices), and severe hypercatabolic symptoms such as cachexia [53,67]. Splenic irradiation is an alternative to splenectomy in patients with symptomatic splenomegaly. However, while a mild-to-moderate reduction in spleen size and symptomatic relief occur in most MF patients, response is temporary lasting only 6 to 8 months. Furthermore, the risk of prolonged and severe cytopenia is considerable. Therefore, splenic irradiation should be reserved for patients not responsive to conventional treatment [46,53].
JAK inhibitors
Ruxolitinib
The JAK/STAT pathway is aberrantly activated in the majority of patients with MF and is associated with increased levels of proinflammatory cytokines and constitutional symptoms. Ruxolitinib is the first JAK inhibitor approved for patients with intermediate- to high-risk MF by the US Food and Drug Administration in November 2011. In a phase 3 randomized trial (COMFORT-I), 41.9% of patients experienced spleen volume reduction and this response was observed not only in patients who had JAK2 V617F, but also in patients with wild-type JAK2. 45.9% of patients showed significant improvement in symptoms [68]. The ruxolitinib group also displayed a significant reduction in mortality (p = 0.04). Ruxolitinib was generally well tolerated but myelosuppression was frequently observed. Grade 3 or 4 anemia and thrombocytopenia occurred in 45.2% and 12.9% of patients, respectively. Another phase 3 study comparing ruxolitinib with best supportive care (COMFORT-II) also reported similar findings of marked reduction in spleen volume and improvement in MF-related symptoms [69]. Recent updates from COMFORT-II demonstrated ruxolitinib-treated patients had improved survival (hazard ratio, 0.48; 95% confidence interval, 0.28 to 0.85; p = 0.009) [70]. After a 32-month follow-up of a phase I/II trial of 107 patients, ruxolitinib-treated patients showed superior survival compared to historical controls. These data suggest that ruxolitinib may alter the natural course of MF, although longer follow-up is needed. However, whether ruxolitinib can reverse marrow fibrosis or reduce JAK2 V617F allele burden has not been clearly shown.
Ruxolitinib has changed the treatment strategy in MF patients, offering significant and durable improvement in quality of life and splenomegaly. So it is most appropriate for symptomatic patients with intermediate- to high-risk disease who are ineligible for allogeneic hematopoietic stem cell transplantation (HSCT). As its use is limited by anemia, combinations with lenalidomide or pomalidomide are under investigation. Trials of pre-transplant use of ruxolitinib are ongoing to study whether it can improve transplant outcome by reduction of symptoms and splenomegaly and improvement of performance status.
Fedratinib (SAR302503)
Fedratinib is a selective JAK2 inhibitor that demonstrated spleen volume reduction in 47% of patients after 12 cycles in a phase I trial. Side effects included anemia, nausea, and diarrhea. Although a phase III study showed promising outcomes, further development of fedratinib has been halted because of several cases of Wernicke-like encephalopathy [71].
Pacritinib (SB1518)
In a phase I/II trial of pacritinib, 41% of patients achieved spleen volume reduction. Another study in 34 patients showed similar efficacy in spleen volume reduction, and 18% of patients showed complete clinical normalization [72]. Myelosuppression was minimal. Pacritinib is currently being investigated in two phase III trials, PER-SIST-I/II.
Momelotinib (CYT387)
Momelotinib is another JAK1/2 inhibitor. A phase I/II study showed reduction of splenomegaly and symptoms, as well as reduced RBC transfusion requirements [73]. Anemia and spleen response rates were 59% and 48%, respectively, and 70% of transfusion-dependent patients achieved transfusion independency. Improvement was also seen in constitutional symptoms. 25% of patients experienced grade 3/4 thrombocytopenia and the most common non-hematologic adverse effects were lightheadedness and hypotension. Momelotinib is currently undergoing a phase III study comparing it to ruxolitinib.
Emerging therapies
New drugs other than JAK inhibitors are being actively investigated through ongoing clinical trials that have shown variable responses in patients with MF (Table 4).
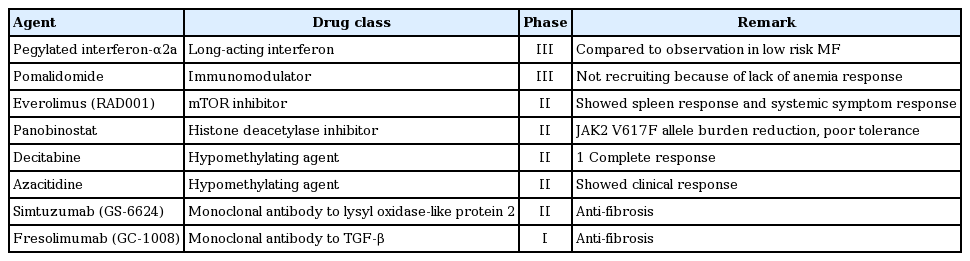
New drugs other than JAK inhibitors for myelofibrosis
Combinations with ruxolitinib
Although ruxolitinib displayed significant efficacy in reducing splenomegaly and symptom burden, it does not reverse all abnormalities in MF. Combination with other active agents may be a reasonable therapeutic approach for MF patients. Lenalidomide, pomalidomide, panobinostat, and monoclonal antibody GS-6624 are under investigation in combination with ruxolitinib in phase II trials (Table 5).

Ongoing combination clinical trials of ruxolitinib
Summary
The approval of ruxolitinib for treatment of intermediate- or high-risk MF based on two phase III trials paved the way to better understanding and further development of treatment options to address the remaining medical needs of MF patients. Ruxolitinib resulted in clinically meaningful relief of splenomegaly and debilitating MF-related symptoms. It also provided significant improvement in survival. However, ruxolitinib is associated with anemia and its effect on marrow fibrosis still needs to be demonstrated. These issues are currently being investigated using combinations of ruxolitinib and immunomodulators, anti-TGF-antibody GS-6624, or pomalidomide. Momelotinib has been reported to alleviate splenomegaly and symptoms and to promote transfusion independency, which needs to be confirmed through controlled clinical trials. It will be quite interesting to see whether the use of JAK inhibitors before allogeneic HSCT improves transplant outcomes in MF patients.
POLYCYTHEMIA VERA
Despite its classification as a neoplasm, the life expectancy of PV patients is not substantially inferior to that of the general population [74,75]. However, patients need to know that vascular disease (thrombosis and hemorrhage) and transformation to either MF or leukemia are the main disease-related complications. Accordingly, prevention of thrombo-hemorrhagic complications and transformation into MF and acute leukemia is the main aim of therapy.
Risk stratification
Thrombosis is the most frequent complication in PV, so the risk stratification system is based on thrombosis risk. According to previous studies, age older than 60 years and history of thrombosis have been identified as major predictors of vascular complications (Table 6) [76,77].

Risk stratification in PV and ET
Response criteria
ELN criteria are used to monitor the response to conventional cytoreductive therapy of PV patients [78]. Criteria for complete response are as follows: hematocrit < 45% without phlebotomy, platelet count ≤ 400 × 109/L, WBC count ≤ 10 × 109/L, normal spleen size on imaging, and no disease-related symptoms.
Clinical management: low-risk patients
Lifestyle modification
To prevent vascular complications, appropriate management of cardiovascular risk factors and maintenance of a healthy lifestyle are important in PV patients, as in the general population. In addition to control of blood pressure, blood glucose, and cholesterol levels, smoking cessation is mandatory.
Phlebotomy
Hyperviscosity secondary to the expansion of blood cells is a major cause of vascular disturbances. To maintain a good ‘blood flow,’ removal of blood (phlebotomy) is a reasonable method. Withdrawing 250 to 500 mL of blood daily or every other day is the usual starting point, and phlebotomy should be continued until a hematocrit between 40% and 45% is obtained [79]. In the elderly or those with a cardiovascular disease, the amount of blood and frequency of phlebotomy may be modified. Although the optimal hematocrit level that should be maintained is still under investigation, the generally accepted level is less than 45% [80]. A recent randomized study by the CYTO-PV Collaborative Group showed that those with a hematocrit target of less than 45% had a significantly lower rate of cardiovascular death and major thrombosis than did those with a hematocrit target of 45% to 50% [81].
Low-dose aspirin
The efficacy and safety of low-dose aspirin (100 mg daily) in PV has been verified in the ECLAP study [82]. After a follow-up of about 3 years, patients on 100 mg aspirin showed a significant reduction in vascular events. Major bleeding was not significantly increased by aspirin. Low-dose aspirin is recommended in all PV patients unless contraindicated.
In summary, low-risk PV patients should be managed with phlebotomy and low-dose aspirin.
Clinical management: high-risk patients
Cytoreductive therapy is indicated in PV patients who are > 60 years old or develop thrombotic or hemorrhagic complications. Otherwise, the administration of cytoreductive therapy may also be justified in low-risk patients who display a progressive increase in leukocyte and/or platelet count, enlarged spleen, or phlebotomy intolerance.
Hydroxyurea
First-line cytoreductive therapy is hydroxyurea. Hydroxyurea is an anti-metabolite that prevents DNA synthesis. The starting dose is 15 to 20 mg/kg/day until response is obtained. Thereafter, a maintenance dose should be administered to maintain the response without reducing the WBC count below 2.5 × 109/L [79]. Supplemental phlebotomy should be performed if needed. Major side effects of hydroxyurea are neutropenia, macrocytic anemia, oral and leg ulcers, and skin lesions.
Interferon α
IFN-α suppresses the proliferation of hematopoietic progenitors and has a direct inhibiting effect on cytokines, which may be involved in the development of MF. Long-term use (median 13 years) of IFN-α in 55 PV patients revealed no thrombohemorrhagic events [83]. IFN-α is not known to be leukemogenic or teratogenic, so it can be used in pregnant women. However, its use in Korea is limited because it is not reimbursed by the NHI program.
In summary, high-risk PV patients should be managed with phlebotomy, low-dose aspirin, and cytoreduction, with either hydroxyurea or IFN-α.
ESSENTIAL THROMBOCYTHEMIA
Risk stratification in ET
Risk stratification of ET is based on assessment of the risk of thrombosis and/or bleeding complication, as the current therapy in ET is aimed at lowering these risks. In the absence of high-risk features, cytoreductive therapy is generally not recommended but could be considered in special cases. In most cases, ET has been associated with a normal life expectancy. True ET is diagnosed according to the 2008 WHO classification [84]. Several new prognostic scoring systems have been proposed, based on additional risk factors such as high leukocyte count (> 11 × 109/L), cardiovascular risk factors, and JAK2 mutational status, but these have not been thoroughly validated [40]. Low-risk patients with extreme thrombocytosis (platelet count > 1,500 × 109/L) are considered separately for bleeding complications [85].
Current recommendations are based on the risk groups delineated in Table 6 [46].
Response criteria in ET
The goal of therapy should be normalized peripheral blood counts in patients who tolerate therapy. At the ELN consensus meeting to develop a definition of response, three different conceptual categories of response were considered: clinicohematologic, molecular, and histologic response (Table 7) [78].
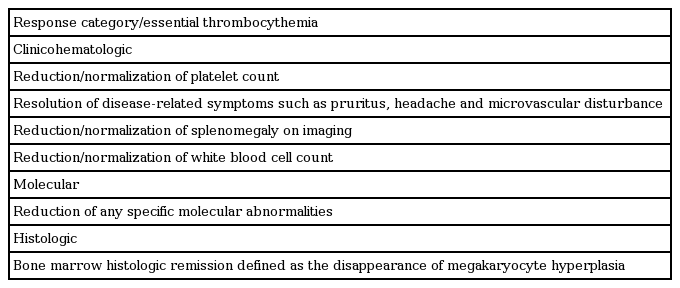
Core set of conceptual criteria to be used for defining response in essential thrombocythemia
Clinicohematologic response definition
At the ELN and IWG-MRT consensus project, new recommendations were based on the concept that standardized criteria for measuring therapy response in ET patients should primarily measure antiproliferative activity and capture long-term effects of new and experimental drugs [86]. The application of these response definitions should be limited to clinical trials, with the understanding that they are not warranted in clinical practice [86]. We recommend the evaluation of clinicohematologic response according to the 2009 ELN criteria in routine clinical practice (Table 8), where the platelet count treatment response was the primary end point. Other parameters, such as disease-related symptoms, spleen size, or WBC count, should be considered along with response.
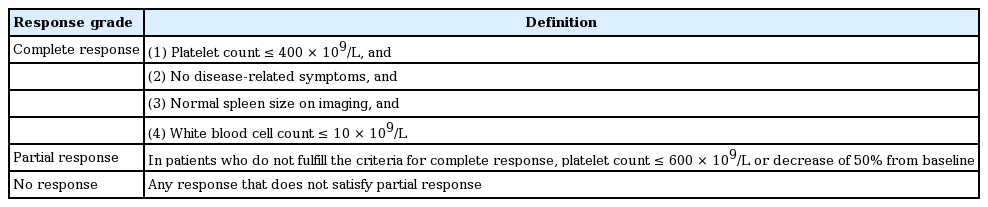
Definition of clinicohematologic response in essential thrombocythemia
Molecular and histologic response
Evaluation of the molecular response, including sequential assessment of the JAK2 V617F allele burden, is not recommended for routine clinical use yet. There is also no indication for repeated BM trephine biopsies in routine response evaluation in ET, but biopsies are essential in assessing the transformation to MF or acute leukemia.
Clinical management of ET
Indications for treatment
Most patients with ET are asymptomatic and can be observed for months or years after the diagnosis is established before requiring treatment. Treatment strategies are based primarily on the presence or absence of risk factors for thrombosis.
Goals of therapy in ET
The goals of treatment are to minimize abnormal clinical, physical, and laboratory features, while also minimizing long-term complications of the disease.
Clinical management: low-risk patients
Young patients with ET (age < 60 years) with no history of thrombosis or hemorrhage and a platelet count < 1,500 × 109/L were prospectively followed and evaluated for thrombohemorrhagic events. The incidence of thrombosis in the study patients (1.9/100 patient-years) was not significantly different from that of an age- and sex-matched control population (1.5 cases/100 patient-years) [87]. These data support the practice of observation without cytoreductive therapy in young, low-risk asymptomatic patients with ET, including those with extreme thrombocytosis [88]. The use of low-dose aspirin (40 to 100 mg/day by mouth) in low-risk patients is encouraged in the presence of vasomotor symptoms or general indications for aspirin use.
Low-risk patients with high platelet counts
It is reasonable to consider cytoreductive therapy in patients with extreme thrombocytosis. It is equally acceptable to manage those patients who do not have acquired von Willebrand disease by clinical observation and/or low-dose aspirin, as long as they avoid tobacco and doses of aspirin > 100 mg/day.
Clinical management: high-risk patients
The risk of recurrent thrombosis is unacceptably high in patients with a previous history of thrombosis or age > 60 years. In a randomized study involving 114 ET patients, the use of hydroxyurea, compared with no treatment, was shown to reduce the risk of thrombosis in high-risk patients from 24% to < 4% at a median follow-up of 27 months [89]. This benefit was maintained after a median follow-up of 73 months [90]. In a second trial, 809 ET patients were randomly assigned to receive treatment with either hydroxyurea or anagrelide, both in combination with aspirin. This trial demonstrated the superiority of hydroxyurea plus aspirin, in terms of prevention of both thrombosis and transformation into MF [91]. Accordingly, we and others recommend the use of hydroxyurea and aspirin as first-line therapy in high-risk ET patients.
Because of its toxicity profile, we do not recommend IFN-α in ET patients except for cases of either treatment failure with other agents or pregnancy. It is reasonable to consider the use of radioactive phosphorus in older patients with an anticipated life-expectancy of < 10 years. In all other settings, we use hydroxyurea as first-line therapy. The recommended initial dose of hydroxyurea is 15 mg/kg per day by mouth, taken in divided doses. Doses are then adjusted up or down depending upon the balance between the desired effect on the platelet count, which should be kept in the range of 100 to 450 × 109/L, and undesired effects such as neutropenia and anemia.
Smoking
Smoking is probably thrombogenic in ET [89,92] and has been reported to alter the inhibition of in vivo platelet activation by low-dose aspirin [93]. This should be emphasized to patients, especially if they wish to profit from the possible benefit of low-dose aspirin [94,95]. We do not recommend cytoreductive therapy in ET based solely on the presence of smoking or obesity; these risk factors should be managed by lifestyle modifications.
Pregnant women or those who desire to become pregnant
It cannot be concluded that specific therapy influences the outcome of pregnancy in low- or intermediate-risk ET patients. We recommend observing these patients without specific therapy.
The use of a platelet-lowering agent may be necessary in high-risk women. Currently, both hydroxyurea and anagrelide are contraindicated for use during pregnancy. On the other hand, several successful pregnancies have been reported in ET patients receiving therapy with IFN-α [96-99]. However, the use of anagrelide or hydroxyurea should not be withheld from high-risk pregnant women who do not tolerate IFN-α therapy, provided well-informed signed consent is obtained.
CONCLUSIONS
Our improved understanding of the molecular pathogenesis of MPNs has prompted the development of targeted agents. The JAK1/2 inhibitor, ruxolitinib, was the first JAK inhibitor approved for patients with intermediate- to high-risk MF. Currently, many kinds of molecular-targeted agents are under investigation. Therefore, we hope to control MPNs effectively and safely in the future with new therapeutic options.
Notes
No potential conflict of interest relevant to this article was reported.