


To the Editor,
Henoch-Schonlein purpura (HSP) is a generalized vasculitis characterized by manifestations of the skin, joints, gastrointestinal tract, and renal involvement. Although HSP can occur at any age and is usually a self-limited disease in children, it is less common and has a poor prognosis in adults. Renal involvement affects approximately one-third of HSP patients, and varies from intermittent hematuria and proteinuria, to severe nephrotic-nephritic syndrome with kidney histology being identical to that of immunoglobin A (IgA) nephropathy.
Fabry disease is an X-linked glycosphingolipid disorder caused by deficient activity of the enzyme α-galactosidase A (a-GLA), which results in the systemic accumulation of globotriaosylceramide (Gb3) in all tissues of the body, including the skin, cornea, heart, and kidneys. The coexistence of Fabry disease and IgA nephropathy has been previously described [1-5].
Here, we report a case of Fabry disease previously diagnosed as HSP. A 45-year-old male was admitted to the clinic with abdominal pain and hematochezia. A gastroscopy and colonoscopy revealed an active duodenal ulcer and colitis, respectively. The patient’s medical history revealed a pontine hemorrhage and hypertension. He had also been consulting a dermatologist for 3 years for petechiae and keloids on his legs and upper arms. The patient was a carrier of hepatitis B for the past 5 years, and had been undergoing treatment with entecavir (0.25 mg) for the past 4 months. The patient had a family history of hepatitis B, and quit smoking and consuming alcohol after his pontine hemorrhagic event. He had hematuria (10 to 29 red blood cells/high power field) and proteinuria (1,582 mg/day), and his serum creatinine level was 1.8 mg/dL. Immunofluorescence microscopy of a renal biopsy (Fig. 1) revealed mesangial IgA deposition, and electron microscopy revealed laminated structures mimicking zebra bodies that had not been recognized at the initial observation. The patient was diagnosed with HSP and prescribed prednisolone (30 mg/day) and eprosartan (600 mg/day). After 2 months, the patient’s serum creatinine and urinary protein levels were 1.9 mg/dL and 1,262.2 mg/day, respectively, and his prednisolone dose was tapered to 10 mg every other day.
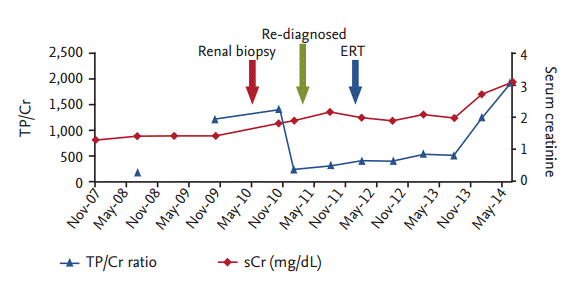
One year later, the patient’s serum creatinine level increased to 2.5 mg/dL and his urine total protein/creatinine (TP/Cr) ratio was 352 mg/g. The initial renal biopsy was reviewed by another pathologist and nephrologist, and the patient was evaluated for genetic mutations and a-GLA enzyme activity. The results showed that the patient had a GLA mutation on the Xq22 gene, and lymphocytic α-GLA activity of 4.4 nmol/hr/mg (reference range, 25 to 126). The patient’s brother and sister underwent an α-GLA examination and the activities were 2.2 and 31.6 nmol/hr/mg, respectively. Following the additional examinations, the patient was diagnosed with Fabry disease and supervening HSP, and began enzyme replacement therapy (ERT) with recombinant human α-GLA. Twelve months following ERT, the patient’s serum creatinine and urinary TP/Cr were 2.0 mg/dL and 541 mg/g, respectively (Fig. 2).
The incidence of Fabry disease has been estimated at 1 in 40,000 to 1 in 60,000 males; however, the true incidence is thought to be higher, as patients with milder forms of the disease are not always diagnosed. Diagnosis of Fabry disease is confirmed by a low α-GLA activity in leukocytes or plasma. It is also diagnosed by a renal histology showing Gb3 accumulation in the kidneys, and lipid inclusions appearing as small, dense granules and larger, complex, zebra bodies using electron microscopy. Life expectancy is considerably shortened in males with Fabry disease, where cardiovascular disease is the most common cause of death. The accumulation of Gb3 in the kidneys leads to a decline in renal function and typically progresses to end-stage renal disease, requiring hemodialysis and/or kidney transplantation in the third to fifth decades of life. Patient survival on dialysis is typically 41% at 5 years. In a recent study, ERT improved renal function and proteinuria, and additional studies have suggested that ERT has metabolic, morphologic, and clinical efficacy against extra-renal manifestations. Histologic examinations of renal, cardiac, and skin cells have shown a complete or partial clearance of Gb3 using ERT, with the greatest benefit occurring in patients who began treatment early in life.
Since Yoshida et al. [1] reported Fabry disease associated with IgA nephropathy in 1994, five more cases have been identified (Table 1) [1-5]. However, there has been no report of Fabry disease combined with HSP. Although the prevalence of IgA nephropathy is highest in Asia, it is not clear if IgA nephropathy is concomitant with Fabry disease, or that Fabry disease causes mesangial IgA deposition. Whybra et al. [4] and Shimohata et al. [5] suggested that aberrant glycosylation of the IgA1 hinge region caused IgA nephropathy in Fabry disease. This hypothesis would be supported by the disappearance of IgA in the patient’s mesangial areas after ERT, or the presence of IgA nephropathy in the renal biopsies of the patient’s siblings; however, the patient’s siblings did not undergo a renal biopsy prior to beginning ERT.
In conclusion, we report a rare case of Fabry disease that was previously diagnosed as HSP. Based on this study, we suggest that clinicians perform genetic and enzymatic activity studies prior to reaching a final diagnosis in similar cases.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





