


To the Editor,
Rosai-Dorfman disease (RDD) was first described by Rosai and Dorfman [1] in 1969 including four cases. In 1990, 433 cases with massive lymphadenopathy were described. RDD is a rare histiocytic proliferative disorder that occurs in children and young adults that mean age at the time of diagnosis is 20.6 years. Patients commonly present with massive, painless lymphadenopathy, fever, malaise, night sweats, neutrophilia, high erythrocyte sedimentation rate (ESR), and polyclonal hypergammaglobulinemia [2].
Extranodal manifestation is common and seen in up to 43% of patients. The skin is the most common extranodal site of involvement (27%) followed by the soft tissue of the nasal cavity. Only 15% of patients have splenic abnormalities. Furthermore, most of them have only mild splenomegaly on physical examination. Only a single case of splenic involvement with hypereosinophilic syndrome has been reported [3]. There are no literature regarding the histologically proven isolated spleen and liver involvement without lymphadenopathy in RDD.
The etiology and pathogenesis of RDD are unclear. The hypotheses include immune regulation disorder or infection, such as various herpes viruses, Epstein-Barr virus (EBV), cytomegalovirus (CMV), Brucella, or Klebsiella. Several infective agent or antigen can cause proliferation of histiocytes, and the key factor is thought to be macrophage colony-stimulating factor. Lymph node histology shows clonal histiocytes in all forms and the massive sinus infiltration of histiocyte-macrophage cells. It has a positive staining for S-100, alpha-1 antichymotrypsin, and CD68 antigens while negative staining for CD1a [4].
Many cases are self-limiting and have indolent clinical courses. There is no ideal treatment for the disease. One-half of the patients have spontaneous resolution. Surgery is a treatment modality for symptomatic disease such as airway compression or nasal obstruction. Multiorgan involvement or association with immune dysfunction is an indication of systemic treatment.
Several modalities of treatment have been used, such as radiotherapy, chemotherapy, and glucocorticoid therapy. There is no standard chemotherapy regimen; however, the most common chemotherapeutic agents are methotrexate, 6-mercaptopurine, and cladribine or vinorelbine plus methotrexate. Interferon treatment has been reported also [5]. The progress of the disease is variable. Regular follow-up is important in the management of these patients.
A 58-year-old woman admitted to be evaluated for anemia in May 2013. The patient presented with dyspnea and cramping pain in both hands, which lasted for one month. She did not have fever, weight loss, night sweats, myalgia, or arthritis. She is never smoked, social drinker, and was not on any medication except for an iron supplement. A physical examination revealed multiple purple bruises and hepatosplenomegaly without peripheral lymph node enlargement.
A blood laboratory test revealed anemia of 4.7 g/dL that had decreased rapidly from 7.8 g/dL 1 month prior. The reticulocyte fraction was 10.2%. The platelet count was severely decreased to 22,000/╬╝L, and it was refractory to repeated transfusions. A peripheral blood smear showed macrocytic hypochromic red blood cells, anisocytosis, polychromasia, poikilocytosis, and teardrop cells. Iron, vitamin B12, and folate measured 40 ┬Ąg/dL (normal range, 50 to 130), 159 pg/mL (normal range, 200 to 950), and 8.12 ng/mL (normal range, 3 to 17), respectively. Total bilirubin was slightly elevated at 1.9 mg/dL (normal range, 0.2 to 1.2). ESR was 5 mm/hr (normal range, 0 to 20). Human immunodeficiency virus antigen/antibody and serologic test for hepatitis were negative. EBV antigens were also negative. CMV, herpes virus, bacterial test, and immunoglobulin in serum were not performed initially.
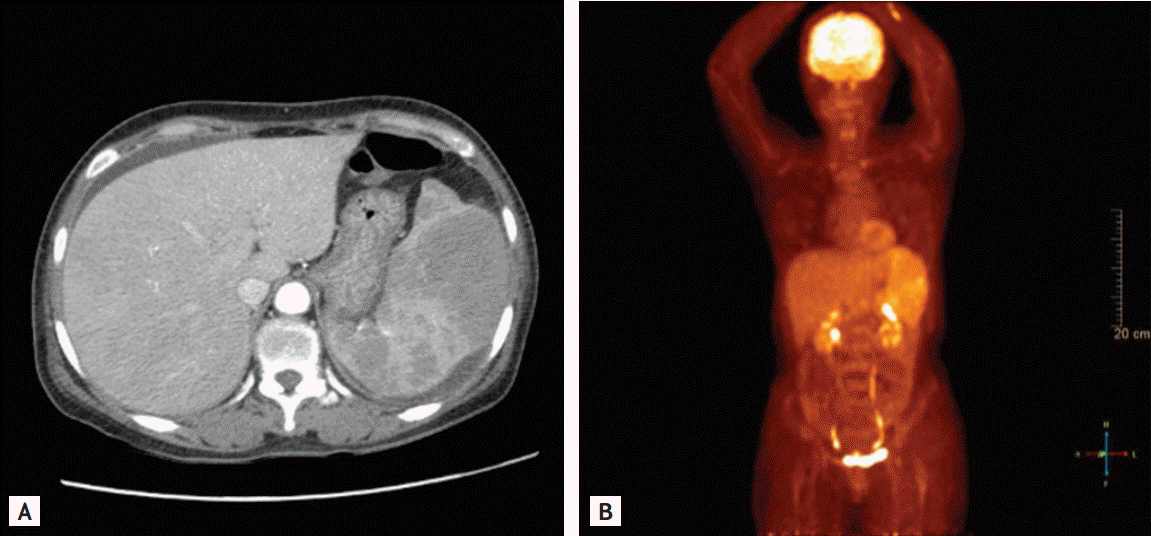
Abdominal computed tomography showed hepatosplenomegaly with heterogeneous enhancement of the liver and multiple nodules of the spleen. Abnormal lymphadenopathy was absent (Fig. 1A). On bone marrow aspiration and trephine biopsy, hypercellular marrow with reactive erythroid hyperplasia was observed. Fluorodeoxyglucose-18 positron emission tomography revealed no hypermetabolic lesion except for an enlarged spleen with diffuse hypermetabolism (Fig. 1B). Magnetic resonance imaging of the brain was normal.
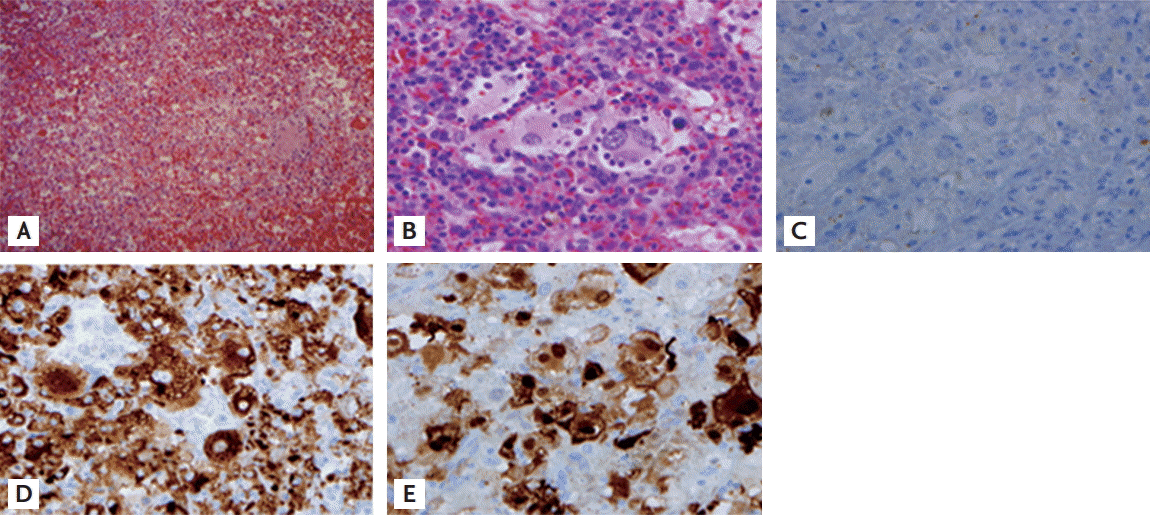
The patient underwent splenectomy. The cut surface revealed multifocal yellowish-brown or mahogany-colored patchy lesions without well-defined masses. On low-power histologic examination, the spleen showed a multifocal hemorrhagic infarct with diffuse infiltration of pale-staining plump cells. These large histiocytic cells were characterized by emperipolesis, the engulfment of intact lymphocytes or red blood cells (Fig. 2A and 2B).
Immunohistochemically, these cells were negative for CD1a (Fig. 2C) and positive for CD68 (Fig. 2D) and S-100 (Fig. 2E), which was consistent with RDD. EBV was not detected in the specimen by Epstein-Barr encoding region in situ hybridization. Hematologic deterioration due to hypersplenism, such as hemolytic anemia with reticulocytosis and thrombocytopenia, dramatically improved after splenectomy.
In the current case, we presented a rare case of RDD that showed only spleen and liver involvement without lymphadenopathy. Splenectomy alone could be both a diagnostic and therapeutic approach in such a patient.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





