


INTRODUCTION
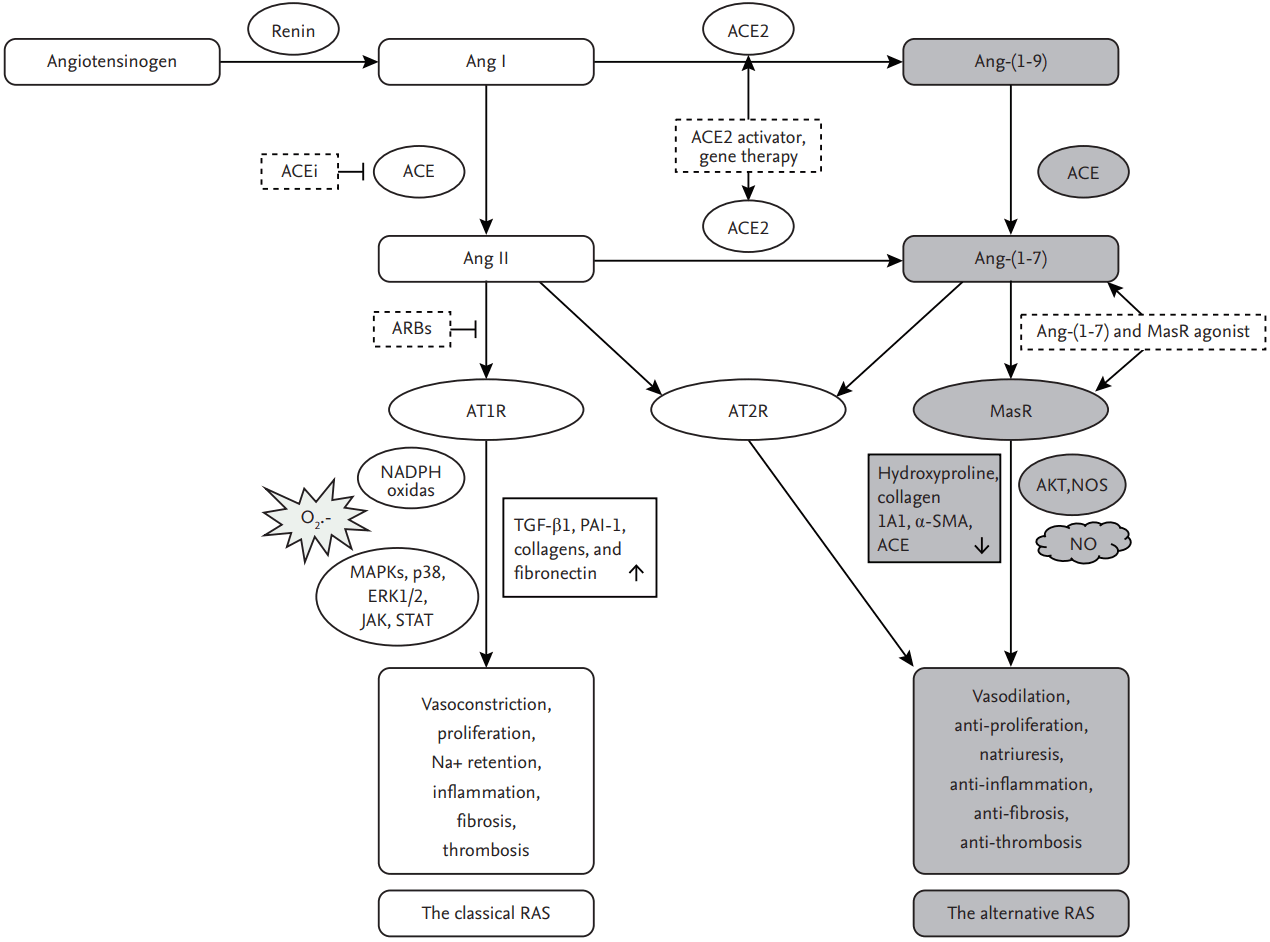
The renin-angiotensin system (RAS) is a physiological regulator of blood pressure, electrolyte balance, and fluid homeostasis. However, it is also involved in organ dysfunction and chronic tissue damage, via the vasoactive and profibrotic effects of angiotensin (Ang) II, a major effector octapeptide [1], and in the pathogenesis of hepatic fibrosis and portal hypertension [2-4]. This review provides an overview of the RAS, its role in hepatic fibrosis and portal hypertension, and current therapeutic approaches based on the use of RAS antagonists to treat patients with portal hypertension (Fig. 1).
OVERVIEW OF THE RAS
The RAS precursor angiotensinogen and its cleavage enzyme, renin, have been extensively detected in normal and in injured liver tissue, but their expression does not increase immediately after liver injury [5]. Instead, the levels of angiotensin-converting enzyme (ACE) and Ang II type 1 receptor (AT1R) are remarkably increased after liver injury, particularly in fibrotic areas of the injured liver tissue and activated hepatic stellate cells (HSCs) [5-7]. ACE2 is expressed in healthy liver by endothelial cells, bile duct cells, and perinuclear hepatocytes, but its high level of expression in the parenchymal tissue of diseased livers results in increased expression of Ang-(1–7), produced from Ang II [8,9]. One cause of portal hypertension is an increase in intrahepatic resistance to portal flow, mediated by contraction of the sinusoidal vascular bed by HSCs and vascular smooth muscle cells [10-12]. HSC activation is facilitated by Ang II. These results link the RAS to liver fibrosis and portal hypertension [2-5,13,14], but they also indicate that the RAS can be targeted to ameliorate hepatic fibrosis. This has been demonstrated in experimental animal models and suggests that a reduction in portal pressure in humans can be achieved by suppressing Ang II-mediated intrahepatic vasoconstriction [15-18].
As the main effector of the RAS, the potent vasoconstrictor Ang I regulates arterial blood pressure and sodium homeostasis, but it also acts on cardiovascular processes such as remodeling [19]. The aspartyl protease renin, expressed by the juxtaglomerular apparatus of the kidney, converts liver-derived angiotensinogen into Ang I, which in turn is converted to Ang II by ACE. ACE2 catalyzes the conversion of Ang II to Ang-(1–7). Thus, Ang II levels are regulated by the balance between ACE and ACE2.
The various components of the RAS form two distinct pathways: the classical pathway, comprising Ang II, ACE, and AT1R, and the alternative pathway, comprising ACE2, Ang-(1–7), and Ang-(1–7) Mas receptor (MasR).
Classical RAS pathway
Vasoconstriction, sodium homoeostasis, fibrosis, cell proliferation, and inflammation are mediated by the classical RAS pathway [20-24]. The vasoconstrictor effect of Ang II is dependent on AT1R, a G-protein-coupled receptor present in most cells types, including the liver [25,26]. AT1R stimulates the activation of phospholipases A2, C, and D as well as L-type Ca2+ channels while inhibiting adenylate cyclase (reviewed in [27,28]). Triggered by Ang II, AT1R also induces cell proliferation by activating tyrosine phosphorylation, phospholipase C-γ, and downstream proteins, including mitogen-activated protein kinases (MAPKs), Janus kinases, and signal transducer and activator of transcription [23,29,30].
In addition to the classical RAS pathway, the (pro)-renin receptor, the ligands of which are renin and pro-renin, activates the extracellular signal-regulated kinase (ERK) 1/2 and p38 pathways, leading to up-regulation of profibrotic genes, including those encoding transforming growth factor β1 (TGF-β1), plasminogen activator inhibitor-1, collagens, and fibronectin [31,32].
Alternative RAS pathway
The alternative RAS pathway, via its components ACE2, Ang-(1–7), and MasR, counterbalances the effects of the classical RAS pathway. Despite the > 40% homology between the catalytic domain of ACE and that of ACE2 [33,34], the latter removes a single amino acid at the C-terminus of Ang I to generate the nonapeptide Ang-(1–9), whereas ACE digests a dipeptide to convert Ang-(1–9) to Ang-(1–7) [34,35]. ACE2 also cleaves Ang II to produce Ang-(1–7), with greater efficiency than the conversion of Ang I to Ang-(1–9) [36]. In fact, the substrate preference of ACE2 for Ang II is approximately 400-fold higher than that for Ang I [36]. ACE2 is thus representative of a compensatory pathway in the RAS [34,37]. Ang-(1–7) and Ang-(1–9), produced by ACE2, react with MasR and AT2R, respectively [38]. MasR is a G-protein-coupled receptor encoded by the Mas proto-oncogene [38]. The ACE2/Ang-(1–7)/MasR axis counterbalances the effects of the ACE/Ang II/AT1R axis, increases vasodilation and the production of nitric oxide (NO), inhibits cell proliferation and cardiovascular remodeling, and improves endothelial function [39,40]. The ACE/Ang II/AT1R axis is also countered by the ACE2/Ang-(1–9)/AT2R axis, which stimulates vasodilation and exerts antigrowth, proapoptotic, and anti-inflammatory effects [41,42].
THE RAS IN HEPATIC FIBROSIS AND PORTAL HYPERTENSION
The hallmarks of cirrhosis are fibrotic septa, regenerating hepatocyte nodules, hepatic sinusoidal remodeling, and capillarization [43,44]. These architectural changes are related to increased intrahepatic resistance to portal blood flow, resulting in increased portal pressure and the development of portal hypertension, the most serious complication of cirrhosis [45]. Portal blood flow is also increased by splanchnic vasodilatation and hyperdynamic circulation [46]. Paizis et al. [5] demonstrated that ACE and AT1R levels are dramatically upregulated in the active fiber-forming region of the liver after hepatic injury; thus, linking the classical RAS to portal hypertension. In addition, ACE and AT1R are highly expressed by activated HSCs both in vivo and in vitro [6]. Upregulation of ACE2 at the gene and protein levels following liver injury in rats and humans implicates the alternative RAS in the response to cirrhosis and portal hypertension [8]. Herath et al. [9] reported the association of alternative RAS activation in chronic liver injury, based on the increase in plasma Ang-(1–7) induced by the upregulation of ACE2 and Mas as well as the hepatic conversion of Ang II to Ang-(1–7). These results clearly show that the classical RAS pathway promotes, while the alternative pathway antagonizes the progression of cirrhosis and portal hypertension.
Role of the RAS in hepatic fibrosis
Sustained and chronic liver disease, caused by hepatitis viruses, heavy alcohol use, certain medications, toxins, and autoimmune diseases, is characterized by the accumulation of excess extracellular matrix (ECM) proteins and changes in liver architecture, followed by the formation of fibrous scars and cirrhotic nodules [2]. Portal fibroblasts, circulating fibroblasts, and bone marrow-derived cells are involved in hepatic fibrogenesis [47], but the most pivotal cell type is HSCs, which secrete collagen types I and III [2]. One of the many mechanisms underlying activation of quiescent HSCs after liver damage is upregulation of RAS components during liver disease, including AT1R/AT2R and MasR, which promote and suppress fibrosis, respectively [5,9,48]. In human liver, quiescent HSCs do not express RAS components, nor do they release Ang II. However, both in vivo-activated HSCs isolated from human cirrhotic liver and culture-activated HSCs isolated from normal human liver highly express active renin and ACE and secrete Ang II [6]. Acting via AT1R, Ang II stimulates DNA synthesis and increases the contraction and proliferation of activated HSCs [49]. Ang II also mediates the proliferation and contraction of HSCs as well as their production of ECM via different signaling pathways, including MAPK pathways, phosphoinositide/Ca2+ pathway, and the generation of reactive oxygen species by phosphorylating the p47phox subunit of Nox [49-51]. HSCs are activated by reactive oxygen species, whereas fibrosis after liver injury is ameliorated in p47phox knockout mice [50]. In addition, in both activated and quiescent rat HSCs exposed to Ang II, the mRNA and protein levels of all TGF-β isoforms are upregulated via the ERK1/2- and Nox-dependent pathways, but independently of protein kinase C [52].
As described above, the alternative RAS axis produces antifibrotic effects via the components ACE2, Ang-(1–7), and MasR. In a rat model of hepatic fibrosis induced by bile duct ligation, the Ang-(1–7) and MasR agonist AVE 0991 improved fibrosis, reduced the content of hydroxyproline, a major component of collagen, and decreased the expression of collagen 1A1, α-smooth muscle actin, and ACE [53]. These antifibrotic effects were antagonized by pharmacological blockade of the MasR, which induced significant increases in hydroxyproline and total TGF-β1 levels [53-55]. In a mouse model of cirrhosis, ACE2, which is upregulated after liver injury [56], inhibited hepatic fibrosis via destruction of Ang II and production of Ang-(1–7). While the loss of ACE2 activity exacerbates experimental hepatic fibrosis, recombinant ACE2 attenuates hepatic fibrosis in chronic liver injury models, suggesting its therapeutic potential [57].
Taken together, these results demonstrate the important roles played by the classical and alternative RAS pathways in promoting and inhibiting fibrosis, as well as the therapeutic potential of classical RAS pathway antagonists and alternative RAS pathway agonists in patients with hepatic fibrosis.
Role of the RAS in portal hypertension
Portal hypertension is a major cause of morbidity and mortality in patients with cirrhosis. Multiple factors contribute to its pathogenesis, including increased intrahepatic resistance following increased deposition of ECM, distortion of the hepatic vascular architecture [2], and splanchnic vasodilation in response to NO produced by endothelial NO synthase [58-60]. The increased vascular tone and ensuing hepatic resistance to portal inflow have also been attributed to contraction of the sinusoidal vascular bed by activated HSCs and vascular smooth muscle cells [11]. Because the activation of HSCs during liver injury is induced by Ang II, and activated HSCs express Ang II, ACE, and AT1R [5,49], the RAS is a key mediator of the pathogenesis of portal hypertension in cirrhosis [3,61,62].
In addition to increased intrahepatic resistance, the systemic and splanchnic vasodilation that characterizes cirrhosis reflects a hypo-responsiveness to vasoconstrictors such as Ang II, α-adrenergic agonists, and endothelin-1 [63,64]. In contrast to the vasoconstrictor activity of Ang II, Ang-(1–7) is a vasodilator [65] whose systemic levels are dependent on ACE2 activity during the progression of hepatic fibrosis [8,9]. Therefore, increased ACE2 expression may accelerate the transition from vasoconstriction to vasodilation in cirrhosis.
RAS ANTAGONISTS FOR THE TREATMENT OF HEPATIC FIBROSIS AND PORTAL HYPERTENSION
Following the report of Lebrec et al. [66] showing that propranolol, a non-selective β-blocker (NSBB), reduces portal venous pressure in patients with cirrhosis, NSBBs have become a standard treatment in patients with portal hypertension. NSBBs reduce cardiac output and splanchnic blood flow by blocking β-1 and -2 adrenergic receptors, resulting in splanchnic vasoconstriction and decreased portal pressure [67]. These drugs were also shown to reduce the incidence of bleeding (primary prophylaxis) and rebleeding (secondary prophylaxis) in patients with esophageal varices [68,69]. However, NSBBs do not produce optimal responses in all patients; 15% of patients do not tolerate NSBBs, and nearly 50% do not show a therapeutic reduction in the hepatic venous pressure gradient (HVPG < 12 mmHg or a decrease of > 20% from baseline) [70,71]. These resistant patients are instead treated with nitrates, which increase NO levels in the intrahepatic circulation, or with prazosin or clonidine, which inhibit α-adrenergic activity; modest reductions in HVPG were achieved in both groups [72-74].
Recently, very promising hemodynamic results have been achieved using other therapeutic agents that inhibit the RAS (e.g., captopril, losartan, and irbesartan) [75-77]. Decreases in portal pressure in patients with cirrhosis have been obtained with RAS antagonists such as ACE inhibitors (ACEis) and AT1R blockers (ARBs), and aldosterone antagonists (AAs), without adverse events [61,78]. Tandon et al. [61] conducted a systematic review and meta-analysis of 19 controlled trials (678 patients) to evaluate the efficacy and safety of RAS antagonists in reducing portal pressure. The ARBs and ACEis used in the clinical trials included losartan, candesartan, and irbesartan (ARBs) and captopril and enalapril (ACEis) [75-77,79-82]. Captopril effectively reduces portal pressure in patients with portal hypertension characterized by a low portal venous velocity [77]. Thus, captopril and other ACEis may be useful for treating this subset of patients with increased intrahepatic resistance. Encouraging results were also obtained with the AA spironolactone [83,84] and with ARB/ ACEi therapy, in which significant reductions in HVPG compared with a placebo were reported [75,76,79,85]. Although greater reductions in HVPG were achieved with NSBBs than with ARBs/ACEis, the difference according to the pooled individual patient data was not significant [77,80]. The HVPG of Child-Pugh class A patients treated with ARBs/ACEis or NSBBs decreased by 17% and 21%, respectively, whereas there was no significant change in the HVPG of Child-Pugh class B/C patients treated with ARBs/ACEis (3%) [61]. However, the difference in the HVPG between the AA-treated and placebo group was significant. While there were no adverse events in any of these groups according to Tandon et al. [61], individual data obtained from a systematic review and meta-analysis of the efficacy and safety of RAS antagonists in reducing portal pressure suggested a higher rate of adverse events in patients with more advanced liver dysfunction.
The antifibrotic effects of RAS antagonists, including ACEis, ARBs, and AAs, have been evaluated in several clinical studies [82,86-94]. The benefits of candesartan, an ARB, in patients with compensated alcoholic liver fibrosis were first reported as part of a well-established open-label randomized controlled trial (RCT) [82]. Candesartan resulted in significant histological improvements and reduced fibrosis scores, fibrotic areas, and α-smooth muscle actin and hydroxyproline levels [82]. Zhu et al. [94] conducted a systematic review and meta-analysis of RCTs to assess the efficacy and safety of ACEis/ARBs in liver fibrosis. They showed that RAS inhibitor therapy significantly lowered both liver fibrosis scores and the liver fibrotic area in patients with hepatic fibrosis; the good safety profile of these drugs was also demonstrated. Taking these results together, the use of RAS antagonists, such as ACEis, ARBs, and AAs, may decrease portal pressure and ameliorate fibrosis in patients with cirrhosis. However, high-quality RCTs using more accurate evaluation parameters are needed to confirm the effectiveness and safety of RAS antagonists for the treatment of hepatic fibrosis and portal hypertension.
CONCLUSIONS
Portal hypertension is a serious complication of cirrhosis that increases the morbidity and mortality rates of these patients. Several studies have demonstrated the role of RAS in the pathogenesis of hepatic fibrosis and portal hypertension [2-4], based on elevated levels of several RAS components (Ang II, Ang-[1–7], ACE, and AT1R) during the progression of hepatic fibrosis [5,6,8,49]. Acting through the ACE/Ang II/AT1R axis (i.e., the classical RAS), Ang II is the main effector regulating vasoconstriction, sodium homoeostasis, fibrosis, cell proliferation, and the inflammation that accompanies various diseases, including liver cirrhosis [20-22,24]. However, the ACE2/ Ang-(1–7)/MasR and ACE2/Ang-(1–9)/AT2R axes, which make up the alternative RAS, have vasodilatory, antigrowth, proapoptotic, and anti-inflammatory properties that counterbalance the effects of the classical RAS axis to reduce hepatic fibrogenesis and portal hypertension [39- 42,95,96]. Accordingly, the use of classical RAS antagonists (ACEi, ARB, and AA) to treat patients with portal hypertension has been examined, with significant and promising hemodynamic results reported thus far [75,76]. These findings suggest that classical RAS pathway antagonists and alternative pathway agonists are key pharmacological molecules that may offer strategies for treating and preventing chronic liver disease and portal hypertension.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





