


INTRODUCTION
Waldenstr├Čm macroglobulinemia (WM) is a rare lymphoproliferative disorder, with a worldwide incidence of three to five cases per million persons per year [1]. The diagnosis of WM requires immunoglobulin M (IgM) monoclonal gammopathy of any concentration and bone marrow infiltration by lymphoplasmacytic lymphoma (LPL) cells [2]. WM generally occurs in elderly people and follows an indolent clinical course with median survival of 50 to 60 months [3]. Thus, many patients with WM may be asymptomatic at diagnosis, and urgent therapy might not be required in most cases. The Second International Workshop on WM recommended initiating therapy in WM patients with constitutional symptoms, symptomatic lymphadenopathy or splenomegaly, anemia (hemoglobin Ōēż 10 g/dL) or thrombocytopenia (platelet count < 100 ├Ś 109/L), and complications related to increased level of IgM such as neuropathy and amyloid light chain (AL) amyloidosis [4].
Various drugs have been used as primary treatment for newly diagnosed WM, from classical alkylating agents to monoclonal antibodies such as rituximab, and a recent meta-analysis showed that rituximab-based immunochemotherapy could be highly effective for WM, with tolerable toxicities [5]. Furthermore, the use of novel targeted agents such as the Bruton tyrosine-kinase inhibitor ibrutinib improves the outcome of WM patients, according to a recent phase III study comparing ibrutinib and rituximab with rituximab alone [6]. However, early disease progression and death may occur in some patients even though the recent Swedish Lymphoma Registry between 2000 and 2014 showed median overall survival of 96 months [7]. Although they might account for a small portion of WM patients, the identification of patients at high risk of early progression and death is important to prevent treatment failure.
For prognostication in WM patients, the International Prognostic Staging System for WM (IPSS-WM) based on disease parameters evaluated at the time of first-line treatment is the most widely accepted prognostic index, consisting of age > 65 years, hemoglobin Ōēż 11.5 g/ dL, platelet count Ōēż 100 ├Ś 109/L, serum ╬▓2-microglobulin Ōēź 3 mg/dL, and serum monoclonal protein > 7 g/dL [8]. Other prognostic models have been proposed consisting of parameters similar to that of IPSS-WM, such as age or hemoglobin and ╬▓2-microglobulin levels [9-11]. Furthermore, a recent study proposed a progression risk classification of asymptomatic WM (AWM risk) patients using bone marrow LPL cells greater than 70%, increased IgM level higher than 4.5g/dL, albumin less than 3.5 g/dL, and serum ╬▓2-microglobulin Ōēź 4 mg/dL [12]. Thus, we analyzed the feasibility of those risk models to identify patients at high risk of progression and death and explored parameters predicting poor prognosis in WM patients.
METHODS
We reviewed the electronic medical records of patients who were pathologically diagnosed with lymphoma and plasma cell neoplasm at Samsung Medical Center between 2000 and 2018 and searched for the term ŌĆślymphoplasmacytic lymphomaŌĆÖ or ŌĆśWaldenstr├Čm macroglobulinemia.ŌĆÖ Among 72 patients diagnosed with LPL or WM, we identified 55 patients with WM after excluding LPL patients without IgM monoclonal gammopathy. As the purpose of this study was to explore parameters predicting poor prognosis based on baseline clinical and laboratory characteristics in WM patients, we selected only symptomatic WM patients requiring treatment. Thus, we excluded eight patients with asymptomatic WM who did not receive treatment after initial diagnosis. Ultimately, we analyzed 47 symptomatic WM patients and collected parameters at diagnosis known to be related to prognosis, including age at diagnosis, Eastern Cooperative Oncology Group (ECOG) performance status, percentage of bone marrow LPL cells, hemoglobin level, platelet count, serum albumin, ╬▓2-microglobulin, and serum lactate dehydrogenase levels. We further obtained information on presenting clinical manifestations including constitutional symptoms, lymphadenopathy, and hepatosplenomegaly, as well as the site of involvement and presence of AL amyloidosis. Clinical and laboratory characteristics were analyzed, and the best response to the first-line treatment was compared according to the response criteria recommended from the Third International Workshop on WM [13].
We also gathered the results of targeted sequencing from the data registry of our prospective cohort after written informed consent (ClinicalTrials.gov Identifier: NCT01877109). Targeted sequencing was performed with paraffin-embedded tissue samples using the HemaSCAN containing 425 genes related to hematological malignancies (Supplementary Table 1) [14]. Thus, we retrospectively analyzed the mutation profiles of two representative cases to compare the distribution of mutations between good and poor prognoses. Detailed methods have been described previously [14,15]. Briefly, genomic DNA was extracted using a QIAamp DNA Mini kit (Qiagen, Valencia, CA, USA). The mean sequencing coverage was greater than 700x. Somatic alterations including mutations, copy number alteration, and structure variants were called using a previously described pipeline: MuTect version 1.1.6, Lowfreq version 0.6.1, Pindel version 0.2.5a4 software, and a custom-built inhouse algorithm were used [15-17].
For statistical analysis, the chi-square test was used for comparison of characteristics, the KaplanŌĆōMeier method was used for univariate analysis of survival outcomes, and the log-rank test was used for comparisons. Cox regression hazard analysis was also performed for multivariate analysis of overall survival. Overall survival was measured from the date of diagnosis to the date of death from any cause and was censored at the date of the last follow-up visit. Statistical associations were determined by the log-rank test. Two-sided p values < 0.05 were considered significant. All analyses were performed using SPSS version 23.0 (IBM SPSS Inc., Armonk, NY, USA) and R3.6.1 software. This study was approved by the Institutional Review Board of Samsung Medical Center, Seoul, Korea, and the requirement for informed consent was waived because of the retrospective nature of the study (No. 2018-06-149). All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
RESULTS
Characteristics of patients at diagnosis
The median age of the 47 patients was 68 years (range, 27 to 86) at diagnosis, and patients over 65 years old accounted for 62% (29/47) of patients. As all patients were referred from primary physicians or a secondary hospital to our center, a tertiary hospital, most patients had symptoms and/or signs associated with WM, including symptomatic lymphadenopathy, dyspnea, cytopenia, neuropathy, and constitutional symptoms such as weakness. However, the frequency of B symptoms was very low (6%, 3/47). Accordingly, most patients showed good performance status (ECOG PS 0/1, 85%, 40/47). Lymphadenopathy was observed in half of the patients (49%, 23/47), and 16 patients (34%) showed Ōēź 2 involved extranodal sites. Hepatomegaly and/or splenomegaly were found in 38% of patients (18/47), and one other involved extranodal sites included the lung and gastrointestinal tract (Fig. 1A). Clinical manifestations at the time of initial visit to the clinic were variable and included lymph node enlargement, dyspnea, weakness, and peripheral neuropathy (Fig. 1B). The median percentage of tumor cells in bone marrow aspirates was 35% (range, 10% to 90%). More than half of the patients (57%, 27/47) had a hemoglobin level lower than 10 g/dL, while only nine patients had thrombocytopenia (platelet count < 100 ├Ś 109/L, 19%, 9/47). The presence of cold agglutinin was not initially evaluated in most patients, and level of hemoglobin and thrombocytopenia was not significantly associated with percentage of tumor cells in the bone marrow (data not shown). All patients had IgM monoclonal gammopathy, and the level of IgM was variable (median, 3,614 mg/dL; range, 316 to 10,795). However, there were no cases with hyperviscosity symptoms such as headache. Decreased serum albumin (median, 3.6 g/dL; range, 2.3 to 4.4) and increased ╬▓2-microglobulin levels (median, 3.6 mg/dL; range, 1.1 to 9.1) were also observed. Three patients with AL amyloidosis presenting with neuropathic pain or diarrhea were finally diagnosed with WM after bone marrow aspiration and immune phenotyping analysis. However, not all patients received a systemic evaluation sufficient to exclude the presence of AL amyloidosis including biopsy at the time of diagnosis based on the review of medical records. Thus, the exact frequency of AL amyloidosis in WM patients could not be determined by the data of this study.
Treatment and survival outcomes
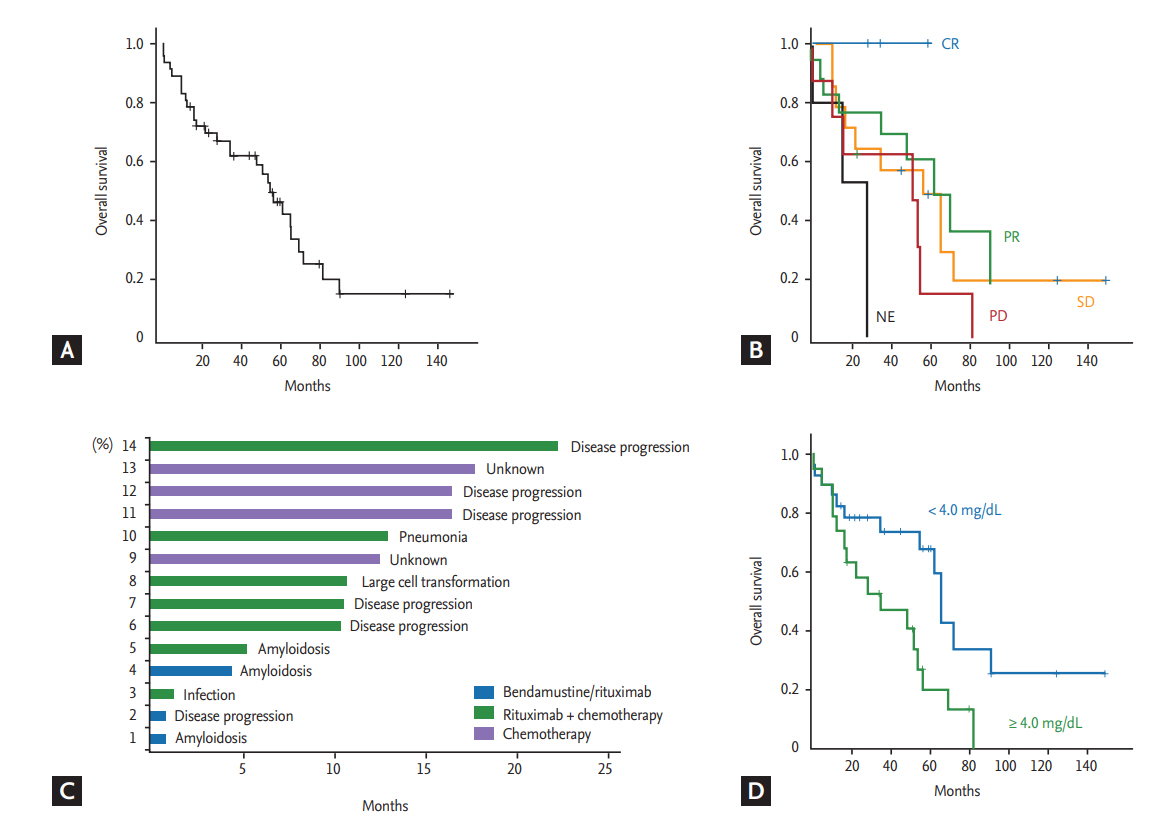
Because use of rituximab was not approved by the Korean Health Insurance System before 2013, only 19 patients received rituximab-containing immunochemotherapy according to previously reported protocols [18,19]: R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone, n = 6), R-CVP (rituximab, cyclophosphamide, vincristine, and prednisone, n = 1), R-CD (rituximab, cyclophosphamide, and dexamethasone, n = 2), or BR (bendamustine plus rituximab, n = 10). The remaining 28 patients who were diagnosed with WM before 2013 received alkylator chemotherapy such as chlorambucil (n = 16) or cyclophosphamide plus prednisolone (n = 12). Out of 10 patients receiving BR, nine responded (complete response [CR] 2, partial response [PR] 7), whereas five patients responded to other rituximab-containing immunochemotherapy (CR1 1, PR 4). Among the 19 patients receiving rituximab-containing immunochemotherapy, there was no case showing laboratory findings suspicious of IgM flare. The response of alkylator chemotherapy was not satisfactory; only six patients showed PR, while the remaining patients showed stable disease (SD; n = 16) or progression (n = 6). With a median follow-up duration of 80.4 months (95% confidence interval [CI], 45.8 to 115.0), 29 patients died due to disease progression (n = 18), organ failure related to amyloidosis (n = 3), infection (n = 4), unknown cause (n = 3), and lung cancer (n = 1). The median OS was 55.1 months at the time of analysis (95% CI, 43.3 to 66.8) (Fig. 2A). Although the number of patients in each treatment group was too small for statistical significance, complete responders to the first-line treatment showed better overall survival (OS) than patients with PR and other responses (Fig. 2B). Among the 47 patients, 14 died within 2 years of the first diagnosis. Their cause of death was mainly associated with disease, including presence of amyloidosis regardless of first-line treatment regimen (Fig. 2C). Patients with amyloidosis who failed to show organ response and clinical symptoms such as pain, diarrhea, and heart failure did not improve, even though serum level of immunoglobulin decreased after chemotherapy. They eventually died due to organ failure related to amyloidosis (Table 1).
Risk factor analysis
Clinical and laboratory characteristics at diagnosis were compared according to the final survival outcome (Table 2). Increased serum ╬▓2-microglobulin level (> 4 mg/dL) wase more frequently found in patients who died compared to surviving patients (p = 0.014). However, there were no other parameters significantly associated with occurrence of death even though we performed statistical analyses using various cutoff values for IgM level, hemoglobin, albumin, and percentage of bone marrow LPL cells. The cutoff of IgM (4.5 g/dL) and bone marrow LPL cells (70%) in the progression risk classification of asymptomatic WM was also not related to the occurrence of death (Table 2). Accordingly, serum ╬▓2-microglobulin level higher than 4 mg/dL was significantly associated with OS (p = 0.015) (Fig. 2D). Among four risk models applied to our patients, IPSS-WM risk and Mayo risk models showed high incidence of death in patients designated as high-risk. However, as 75% of patients belonged to the high-risk group of the Mayo risk model, its clinical relevance was lower than that of the IPSS-WM risk model, designating 43% of patients as high-risk (Table 2). However, the comparison of OS based on risk model showed that only low-risk patients had better OS than intermediate- and high-risk patients (p < 0.05), whereas there was no difference between intermediate- and high-risk patients in the IPSS-WM risk model (Fig. 3A). The association of other risk models with OS was not significant (Fig. 3B-3D).
Mutation analysis
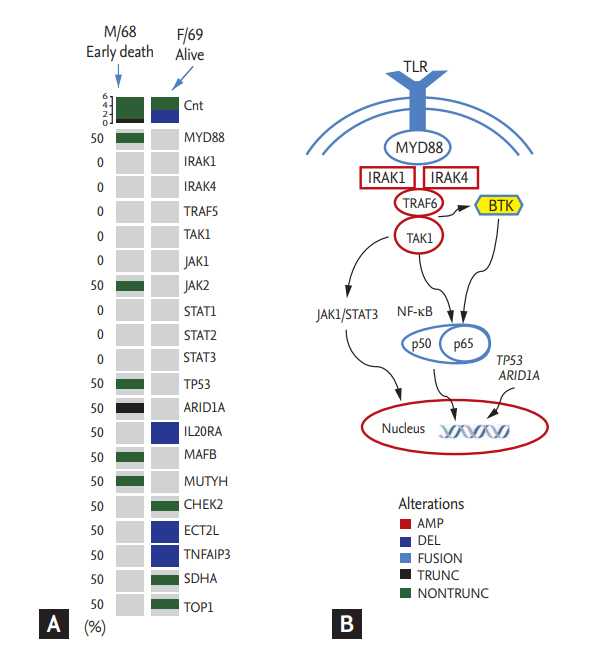
Although evaluation of the MYD88 L265P mutation was not performed in all patients at diagnosis, our previous analysis using the mutant enrichment 3ŌĆÖ-modified oligonucleotide-polymerase chain reaction technique found 21 out of 28 LPL cases (75%) with the MYD88 L265P mutation in bone marrow aspirates [20]. In addition to the above-mentioned 21 cases, we performed targeted sequencing using paraffin-embedded tissue blocks from two representative cases from our study population. One case (male/68 years old with IgM/kappa) showed early death within 2 years after diagnosis. Although he was treated with rituximab-CHOP chemotherapy immediately after diagnosis, disease progression occurred after the fifth cycle and became refractory to subsequent salvage chemotherapies. The other case (female/69 years old with IgM/lambda) survived. Although she experienced relapse 3 years after completion of her firstline treatment (R-CHOP), she responded to the salvage chemotherapy including rituximab. Comparison of sequences revealed differences between the two cases, including mutations of MYD88, TP53, ARID1A, and JAK2 in the ED case (Fig. 4A).
DISCUSSION
WM is an extremely rare disease in Asian countries, and most data are from Western patients. Indeed, a nationwide analysis of the incidence of malignant lymphoma according to the WHO classification between 2005 and 2006 reported an incidence rate of 0.3% in Korea [21]. The clinical course of WM is variable, ranging from asymptomatic cases with increased IgM to symptomatic cases with cytopenia and organomegaly. Thus, approximately 40% of WM patients have a mild form of anemia; other non-specific symptoms may include weakness, fatigue, and weight loss. One-third of patients may have lymph node enlargement and hepatosplenomegaly. As a substantial number of patients with WM follow an indolent course without progression to an aggressive state for a long time, treatment initiation should not be based on serum IgM level. Instead, a ŌĆśwatch and waitŌĆÖ strategy could be considered until patients develop symptoms requiring therapy. However, 29 patients had died at the time of analysis in our study, and the majority was due to disease progression. As 14 patients died within 2 years of diagnosis, the median OS was 55.1 months, which was lower than that of a recently published Swedish nation-wide dataset reporting a median OS of 96 months [7]. This difference might be associated with the symptomatic aggressive WM in the majority of patients in our study, as mentioned above. Furthermore, most patients who were diagnosed with WM before 2013 received chemotherapy with alkylators due to reimbursement issues with rituximab-containing immunochemotherapy. Indeed, their response was poor (PR 6, SD 16, and progressive disease 6), which may have led to inferior outcomes for our patients. Currently, alkylators or nucleoside analogues are not recommended for patients younger than 65 years to avoid secondary malignancies and disease transformation, and rituximab-containing immunochemotherapy regimens have become the mainstay of treatment [22]. In particular, a phase III non-inferiority study comparing BR with R-CHOP as the first-line treatment reported that BR is associated with longer progression-free survival (69 months vs. 29 months) and better tolerance in WM patients [19]. Although we could not show significant difference of OS according to the type of treatment due to the small number of patients in each treatment group and the retrospective nature of our study, the BR regimen could be one treatment option for WM patients like the currently preferred R-CD regimen, considering its efficacy and tolerable toxicity compared to R-CHOP [23].
In this study, we evaluated the predictive value of IPSS-WM and other prognostic models for predicting the poor prognosis in WM patients [8-11]. However, the comparison of OS based on risk model showed that only low-risk patients had better OS than intermediateand high-risk patients, whereas there was no difference between intermediate- and high-risk patients in the IPSS-WM risk model (Fig. 3A). In addition, prognostic values of other risk models were less than we expected in Korean WM patients. When we performed univariate analysis using previously reported prognostic factors, including age more than 65 years, presence of cytopenia, serum IgM level, percentage of bone marrow tumor cells, and poor performance status, only serum ╬▓2-microglobulin level higher than 4 mg/dL was significantly associated with OS. Given that serum ╬▓2-microglobulin level is included as a component of three prognostic models (IPSS-WM, Mayo, and Southwest Oncology Group [SWOG]), measurement of serum ╬▓2-microglobulin might be useful for predicting poor survival outcome of WM as a single biomarker. However, our study has several limitations. First, treatment regimens were heterogeneous, and the number of patients in each treatment was too small to draw a solid conclusion. Second, our results could be influenced by selection bias due to the retrospective nature of this single-institute study. Accordingly, multivariate analysis could not be performed. Further studies with a larger study population should be performed to evaluate the prognostic value of other parameters such as serum IgM level and bone marrow tumor cells considering their potential association with poor prognosis of WM. The increased level of IgM could induce amyloid deposits, resulting in light chain (AL) amyloidosis, and IgM-related amyloidosis is present in 5% to 7% of patients with AL amyloidosis [24,25]. In this study, three patients with AL amyloidosis died due to organ failure related to amyloidosis, even though they all showed a hematologic response to BR or R-CD (Table 1). However, systemic evaluation for the presence of AL amyloidosis was not performed in most patients of this study because amyloidosis is a relatively uncommon event. Thus, the prognostic value of AL amyloidosis in WM patients also should be confirmed in a further prospective study.
As data regarding genomic alterations of WM accumulate, genomics-based prognostication has been tried. Although the MYD88 L265P mutation can be found to a lesser extent in other indolent or aggressive lymphomas such as marginal zone lymphoma and diffuse large B-cell lymphoma, whole-genome sequencing of bone marrow tumor cells reveals MYD88 L265P as a frequent mutation in patients with WM [26]. Better OS has been reported in patients with the MYD88 L265P mutation compared to the MYD88 wild-type [27]. However, the impact of the MYD88 L265P mutation on OS remains controversial, because no association of overall survival with the MYD88 L265P mutation was reported in another study [28]. In our study, not all patients were evaluated for the MYD88 L265P mutation; thus, we could not analyze the association of early death with the mutation. However, the one analyzed case of early death had the MYD88 L265P mutation as well as mutations in TP53 and ARID1A. The TP53 mutation has been observed in 7.3% of WM patients who had shorter survival in a previous study [29]. Truncated mutations of ARID1A have also been reported in WM patients, including single-nucleotide variants leading to premature protein truncation [30]. Although the precise mechanisms by which these mutations influence the occurrence of early death remain to be elucidated, evaluation of mutation profiles at diagnosis might provide helpful information for predicting early death in WM patients, given their crucial role in the pathogenesis of WM (Fig. 4B).
In summary, we analyzed our experience of managing WM patients and evaluated the prognostic relevance of various risk models for WM. Although we analyzed a relatively small number of patients who were heterogeneously treated due to the retrospective nature of the study, our results suggest that serum ╬▓2-microglobulin level and the IPSS-WM risk model can predict poor survival in WM patients. In addition, mutation analysis might provide additional information on risk models based on clinical and laboratory parameters.
KEY MESSAGE
1. Serum ╬▓2-microglobulin level could be a single biomarker strongly predictive of poor survival of Waldenstr├Čm macroglobulinemia (WM) patients.
2. The low-risk group of the International Prognostic Staging System for WM risk model has better prognostic value than other risk models, and mutation analysis might provide additional information to predict a high-risk group.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement 1
Supplement 1 Print
Print





