


INTRODUCTION
Sudden cardiac death (SCD) is a major global health burden [1,2]. A substantial proportion of SCD is related to coronary artery disease and heart failure [1,3]. However, SCD can occur in healthy patients without overt heart disease.4 These non-structural causes of SCD often occur in relatively young patients without prior heart disease and are referred to as inherited arrhythmia (IA) syndrome [3,4]. When appropriate resuscitation is not performed during the acute event, IA can cause grave damage both to the patient and to his or her family. The underlying pathophysiology of IA has not been fully established; however, recent advances in technology for molecular biology and high throughput testing have uncovered the genetic basis for IA syndrome, which has enabled identification and targeting of the culprit substrate [3,5].
IA consists mainly of cardiac channelopathies such as Brugada syndrome (BrS), long QT syndrome (LQTS), short QT syndrome (SQTS), and catecholaminergic polymorphic ventricular tachycardia (CPVT) [3,6ŌĆō8]. Early repolarization syndrome (ERS) and idiopathic ventricular fibrillation (IVF) are other primary electrical disorders. Nonischemic cardiomyopathies, such as hypertrophic cardiomyopathy, dilated cardiomyopathy, or arrhythmogenic right ventricular cardiomyopathy (ARVC), are also genetic disorders that have a risk of SCD.
The IAs have certain clinical characteristics. First, the initial manifestation may be lethal or a severe form like cardiac arrest or syncope due to ventricular tachyarrhythmias. Second, diagnosis of the disease and prediction for SCD may be difficult because of a lack of clinical clues, and structural abnormalities in imaging studies are rare. Third, IA is a genetic or familial disease, and is not curable. Fourth, primary prevention for SCD in clinical practice using an implantable cardioverter-defibrillator (ICD) is still controversial.
In this review, we provide epidemiological, clinical and genetic overviews of IAs that predispose to SCD. In addition, we provide a current update to the clinical approach and genetic testing for IA.
EPIDEMIOLOGY OF SCD AND INHERITED ARRHYTHMIAS
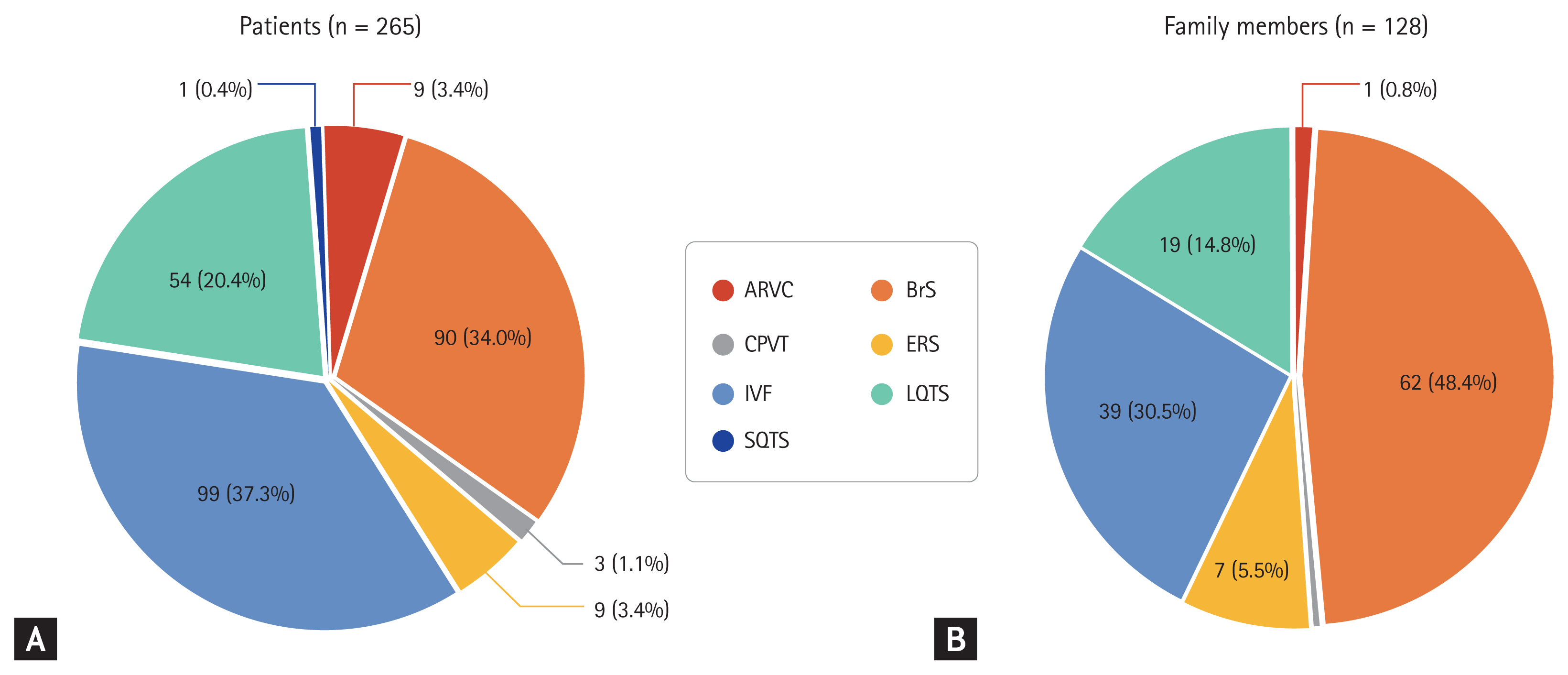
The estimated incidence of SCD in North America and Europe is 50ŌĆō100 cases per 100,000 person-years of follow-up [6,9,10]. The incidence is higher in people older than 35 years with one case per 1,000 person-years of follow-up; however, the risk of SCD is not negligible in people younger than 35 years [11]. SCD can occur not only by ventricular fibrillation (VF) or ventricular tachycardia (VT) but also with pulseless electrical activity or asystole. Furthermore, VF and VT can degenerate into pulseless electrical activity or asystole if left untreated. Estimation of the incidence of IA is made more complicated because substantial proportions of VF and VT are due to structural heart disease such as coronary artery atherosclerosis [12]. According to data from Western countries, coronary artery disease is responsible for 60% to 80% of SCD, while the proportion of SCD with underlying IA is < 5% [13,14]. However, Japanese data indicated that primary electrical disorders related to channelopathies were estimated to be responsible for 10% of SCDs [15]. The proportion of IA in people who experienced SCD in South Korea may be higher than in Western countries and similar to or higher than in Japan. Based on the Korean National Health Insurance Service database, among 1,125,691 people included in the sample cohort, the overall incidence of sudden cardiac arrest was 48.7 per 100,000 person-years, and the rate of sudden unexplained death was 14.7% in patients with SCD [16]. Roh et al. [17] also demonstrated that among patients who received an ICD, identified using the Health Insurance Review and Assessment Service database, the proportion of IA as an etiology associated with the ICD was 21.1%. According to data from the Korean Inherited Arrhythmia Registry, among 265 probands clinically diagnosed with IAs, IVF was the most common (n = 99, 37.3%), followed by BrS (n = 90, 34.0%) and LQTS (n = 54, 20.4%), and among 128 family members, about half were related to a patient with BrS (n = 62, 48.4%), followed by LQTS (n = 39, 30.5%) and IVF (n = 19, 14.8%) (Fig. 1) [18]. The three most common IAs (BrS, IVF, and LQTS) accounted for > 90% of IAs in the probands and their family members in the Korean population.
CLINICAL APPROACH TO INHERITED ARRHYTHMIA SYNDROME
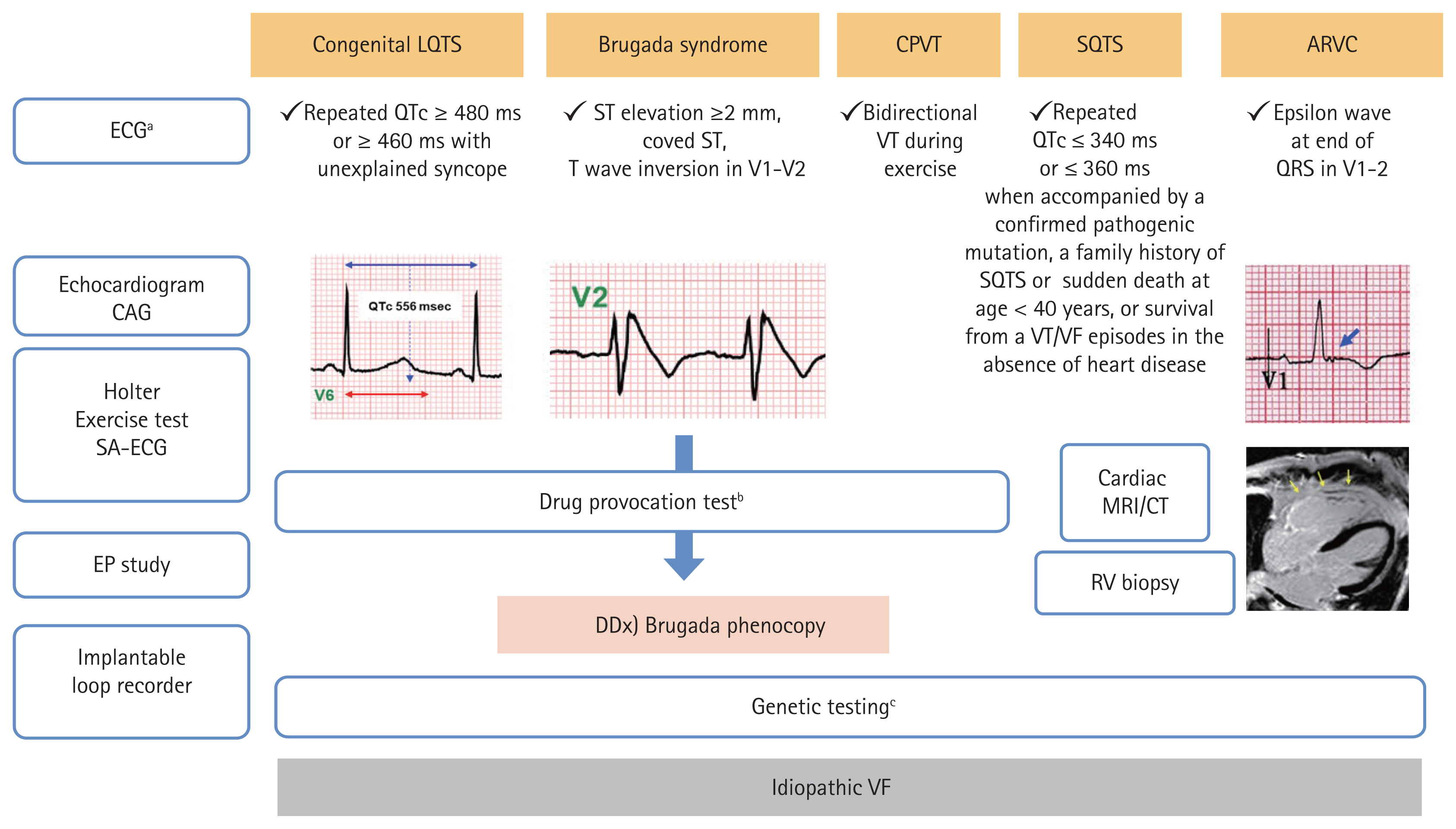
Excitation contraction coupling in the cardiomyocyte is a core process generating heart rhythm, which is mediated by harmonized ionic currents through various ion channels that are imbedded in the sarcolemma. Alterations in the function of cardiac ion channels by genetic mutations can cause electrical heterogeneity and subsequent initiation of ventricular tachyarrhythmias, which are called channelopathies. Gain- or loss-of-function of cardiac ion channels reveal no structural abnormalities of the heart in imaging studies. Major forms of channelopathies responsible for SCD include BrS, LQTS, SQTS, CPVT, ERS, and IVF [19ŌĆō22]. The overall diagnostic approach is summarized in Fig. 2. The underlying genetic pathophysiology and a current clinical update of the channelopathies are reviewed in the following section.
LONG QT SYNDROME
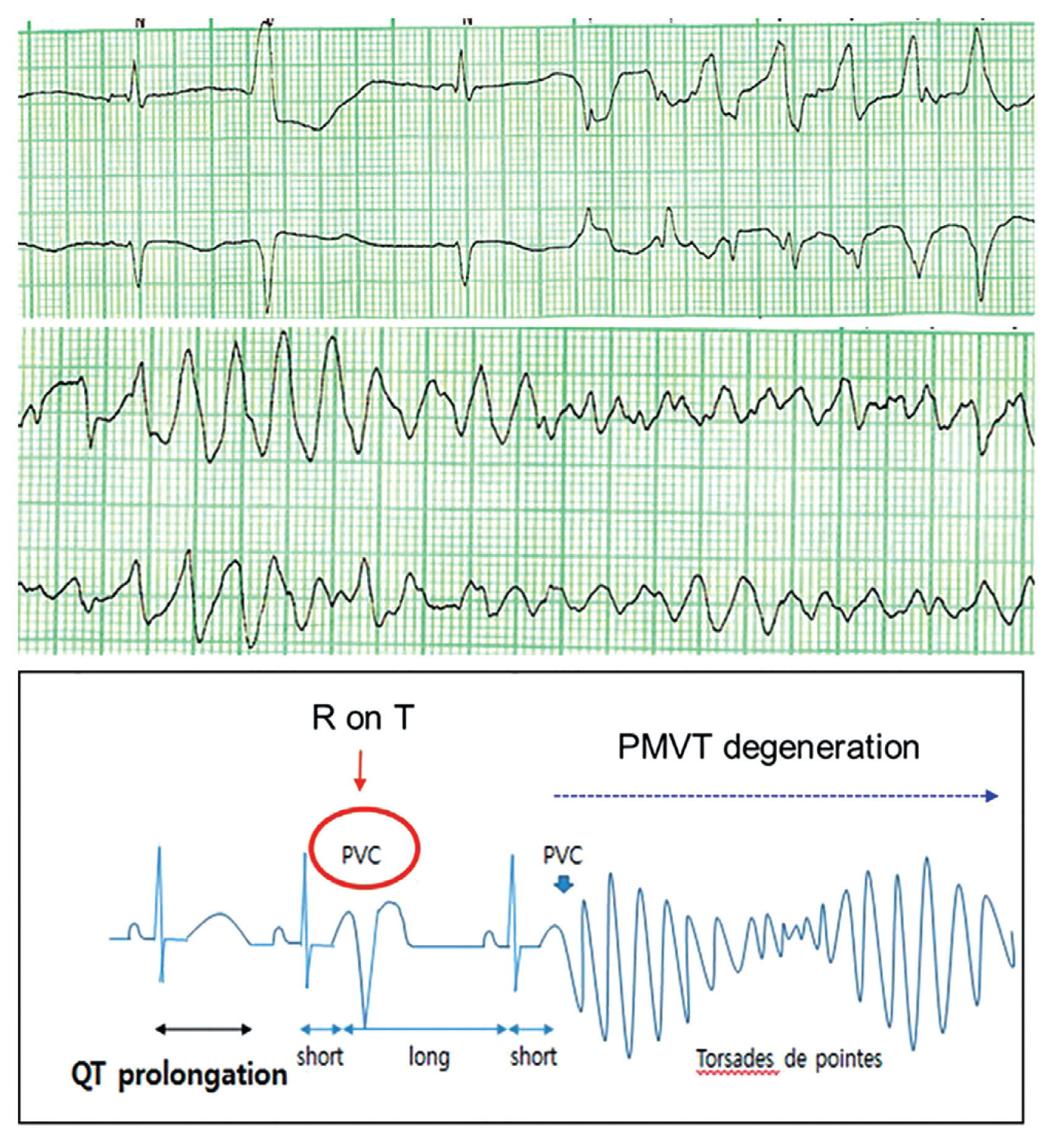
Patients with LQTS experience lethal ventricular arrhythmias accompanied by prolongation of the QT interval reflecting an excessive prolongation in the duration of cardiac repolarization (Fig. 3). An epinephrine challenge test can unmask prolongation of the QT interval in patients with ŌĆśconcealed LQTSŌĆÖ who show a normal range of QT intervals on their resting electrocardiography (ECG). Patients with LQTS are diagnosed by an LQTS risk score > 3, a corrected QT (QTc) interval for heart rate using BazettŌĆÖs formula Ōēź 480 ms in repeated ECGs in the absence of a secondary cause for QT prolongation, or an unequivocally pathogenic mutation in one of the LQTS genes [23]. Among 20 genes that have been discovered, potassium voltage-gated channel subfamily Q member 1 (KCNQ1; LQT1), potassium voltage-gated channel subfamily H member 2 (KCNH2; LQT2), and sodium voltage-gated channel alpha subunit 5 (SCN5A; LQT3) are the most common LQTS genes, accounting for about 90% [3,20,24,25]. Triggers for cardiac events and ECG morphologies that may be characteristic according to the genotypes can assist in management [26]. Affected patients show high mortality rates > 20% within 1 year after the first syncopal episode if left untreated [26,27]. Schwartz reported a significantly reduced mortality in LQTS patients who received beta-blocker or surgical antiadrenergic therapy [27]. Patients with genetic LQTS treated with a beta-blocker experience a high rate of cardiac events, particularly those with LQT2 and LQT3 genotypes [28]. Recent studies have demonstrated the superiority of nadolol, a beta-blocker, in reducing the risk of life-threatening arrhythmic events, suggesting preferential management with nadolol [29,30]. An ICD with the use of a beta-blocker is recommended in LQTS patients with previous cardiac arrest. Left cardiac sympathetic denervation should be considered when a beta-blocker is either not effective, not tolerated or contraindicated, or when an ICD is contraindicated or refused [22]. Table 1 shows the clinical characteristics of LQTS in Korean and international registries [18,31ŌĆō35].
BRUGADA SYNDROME
BrS is an inherited disorder associated with lethal ventricular arrhythmias and results in an increased risk of SCD [36]. BrS is diagnosed in patients with ST-segment elevation with type 1 morphology Ōēź 2 mm in one or more leads among the right precordial leads V1 and/or V2, positioned in the second, third, or fourth intercostal space, occurring either spontaneously or after a provocative drug test with intravenous or oral administration of sodium channel blockers, such as flecainide or procainamide [22]. Brugada phenocopies, which are Brugada-like ECG patterns induced by reversible conditions such as pectus excavatum, should be differentiated from true BrS [37]. Lethal arrhythmias occur predominantly in younger males (20 to 40 years old) during rest or sleep [38]. Among 19 genes known to be associated with BrS, loss-of-function in SCN5A, CACN1AC, and sodium voltage-gated channel alpha subunit 10 (SCN10A) were found individually in > 5% of positively genotyped patients [39]. However, the clinical use of genetic testing results remains limited, which may be due to oligogenic genetic susceptibility and low disease penetrance in BrS compared to forms of Mendelian inheritance [40]. In addition, a subset of patients with BrS also have other types of arrhythmias such as atrial fibrillation or conduction defects such as atrioventricular block [3,41,42]. Lifestyle modification is recommended in all patients with a diagnosis of BrS [23]. Drugs that may induce ST-segment elevation in right precordial leads, excessive alcohol intake, and large meals should be avoided; any fever should be promptly treated with antipyretics. An ICD is recommended in survivors of an aborted cardiac arrest or patients who have documented sustained VT, and should be considered in patients with a spontaneous type I ECG pattern and history of syncope, presumably due to an episode of VT. Table 2 shows the clinical characteristics of BrS in Korean and international registries [18,38,43ŌĆō49].
EARLY REPOLARIZATION SYNDROME
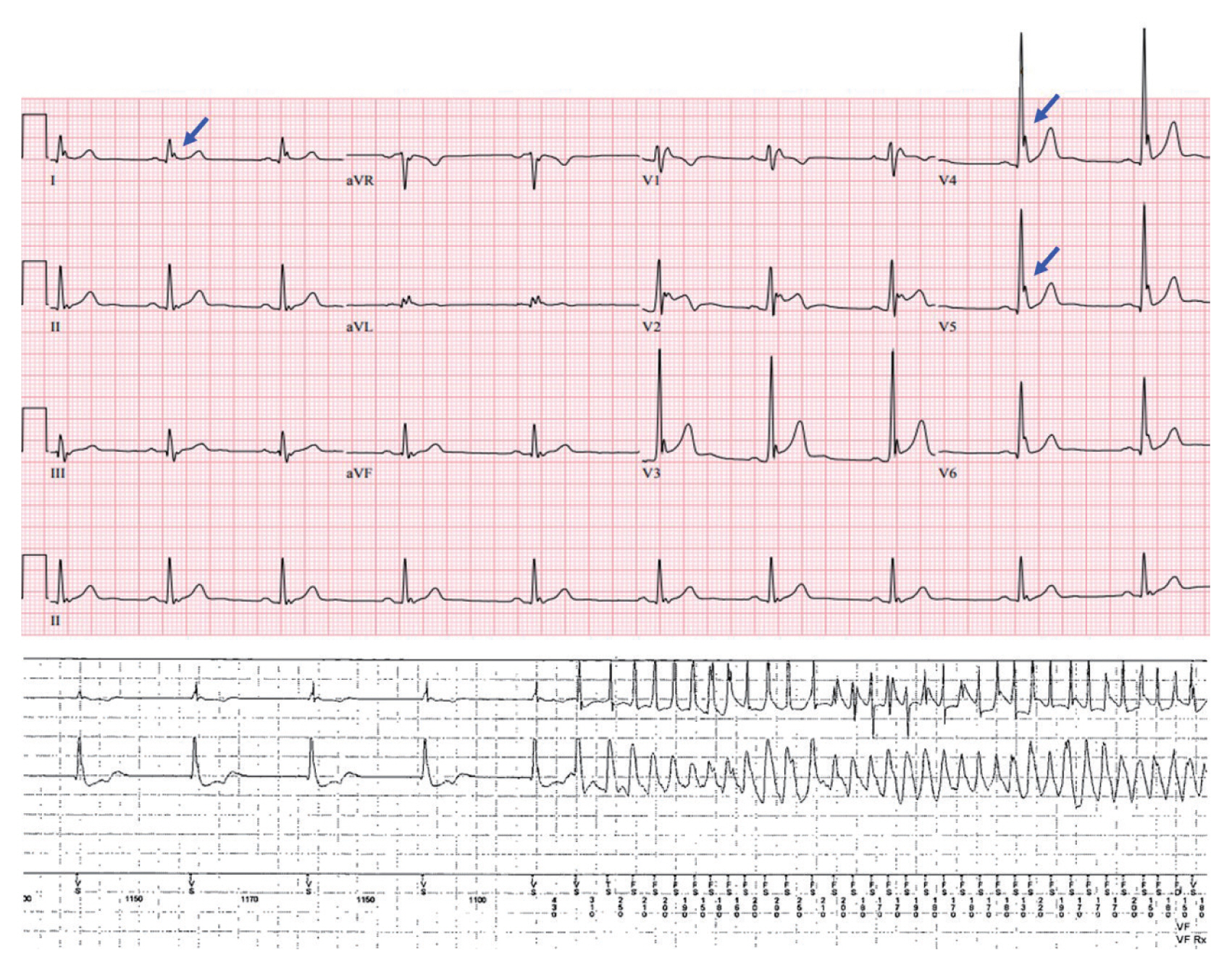
Electrocardiographic characteristics of early repolarization include J-point (the point between the end of the QRS complex and the beginning of the ST segment) elevation, notching or slurring of the terminal part of the QRS complex, and tall and symmetric T waves (Fig. 4) [50]. Previously, early repolarization was considered a benign phenomenon that did not need further evaluation [51,52]. However, the concept has changed since the demonstration that early repolarization in ECG, particularly in the inferior and lateral leads and with greater amplitude, is associated with a significantly increased risk of lethal ventricular arrhythmias and SCD [53ŌĆō55]. The term ERS is used when early repolarization shown in ECG is accompanied by clinical symptoms such as syncope or SCD. The underlying mechanism for J-point elevation and the occurrence of lethal ventricular arrhythmias in ERS has still not been fully elucidated. A voltage gradient that results from transmural differences between the epicardium and endocardium during phases 1 and 2 of the cardiac action potential has been suggested as a possible mechanism of J-point elevation by Antzelevitch et al. [50,56,57]. They also suggested that eliminating the arrhythmia substrate might reduce the future recurrence of lethal ventricular arrhythmias [58,59]. Several novel genetic variations in potassium inwardly rectifying channel subfamily J member 8 (KCNJ8) and SCN5A have been reported to be potential genetic abnormalities responsible for ERS [60,61].
IDIOPATHIC VENTRICULAR FIBRILLATION
IVF is diagnosed when no other cause is identified despite a systematic evaluation including ECG, imaging studies, drug challenge test and genetic evaluation in patients suffering VF. The Heart Rhythm Society/European Heart Rhythm Association/Asia Pacific Heart Rhythm Society expert consensus defines IVF as a resuscitated cardiac arrest victim, preferably with documentation of VF, in whom known cardiac, respiratory, metabolic and toxicological causes have been excluded through clinical evaluation [62,63]. The possibility of IAs or cardiomyopathies should be excluded before making the diagnosis of IVF. Whether IVF is caused by variations in a single or multiple genes is unclear. Interactions between genetic variations and environmental factors such as electrolyte imbalance, sympathetic activities, and undetectable myocardial fibrosis may also play a role [63]. Genetic testing in collaboration with clinical evaluation can facilitate identification of the underlying pathophysiology of IVF. With recent advances in genetic testing and detection of a clinical subset of IVF, the incidence of IVF is expected to decline [63]. Although cardiomyopathies are not part of IA syndrome due to obvious structural abnormalities, a lethal ventricular arrhythmia can be the first manifesting symptom [64]. Evaluating whether these concealed cardiomyopathies are associated with an increased risk of lethal ventricular arrhythmia will evolve with molecular diagnostics. Table 3 shows the clinical characteristics of IVF in Korean and international registries [18,65ŌĆō70].
ARRHYTHMOGENIC RIGHT VENTRICULAR CARDIOMYOPATHY
Arrhythmogenic cardiomyopathy is a genetically determined abnormality of cardiac desmosomes predisposing to SCD, particularly in young patients and athletes. Although involvement is often biventricular, predominant involvement of the right ventricle is the original disease phenotype, hence ARVC. The histopathological hallmark of ARVC is the substitution of right ventricular myocardium with fatty or fibrous tissue [71,72]. Prior studies have revealed that the disease is caused by genetic variations in genes encoding cardiac desmosomes such as junction plakoglobin (JUP), desmoplakin (DSP), plakophilin 2 (PKP2), desmoglein 2 (DSG2), and desmocollin 2 (DSC2) [73ŌĆō77]. Patients with ARVC usually have structural abnormalities such as right ventricular dilation or wall thinning [78]. The diagnosis of ARVC may be difficult due to highly variable clinical presentations [79]. In the early phase, affected patients are at increased risk of lethal ventricular arrhythmias, but show no evidence of right ventricular abnormality and clinical symptoms; this is known as a concealed phase [79]. SCD can be the first clinical manifestation in patients affected by ARVC [80]. Therefore, although ARVC is a cardiomyopathy with structural abnormalities, it can also be called an IA syndrome in which lethal ventricular arrhythmia occurs without obvious structural abnormality in the heart.
Cerrone et al. [81] demonstrated the coexistence of sodium channelopathy and genetic PKP2 variations, suggesting PKP2 mutations may be a molecular substrate leading to the diagnosis of BrS. Accordingly, the overlapping phenotype may be explained by the emerging theory that ARVC and BrS are not completely different conditions, but the ends of a spectrum of structural myocardial abnormalities and sodium current deficiency that share a common origin as diseases of the connexome [82].
GENETIC TESTING FOR INHERITED ARRHYTHMIAS
In the past, the underlying causes of IA were not identifiable due to technical limitations. However, as advances in genetics have improved the understanding of IAs, genetic testing for the channelopathies and cardiomyopathies can have diagnostic, prognostic, and therapeutic implications, potentially influencing clinical decisions (Table 4) [23,43,62,83ŌĆō85]. Recently, the next-generation sequencing technique has enabled multiple genetic variant analysis in a given patient, within a reasonable cost and time requirement. However, not all physicians can incorporate the results of genetic testing into a patientŌĆÖs care, because of potential pitfalls. Unlike solid cancers that are caused by somatic variants, the main outcome measurement in patients with IAs that are mostly caused by germline variants is an event of SCD (i.e., ŌĆśall or nothingŌĆÖ), which is hard to predict. Standards and guidelines developed by the American College of Medical Genetics and Genomics can be used for the interpretation of sequence variants using five specific categories based on evidence: ŌĆśbenign,ŌĆÖ ŌĆślikely benign,ŌĆÖ ŌĆśvariants of unknown significance,ŌĆÖ ŌĆślikely pathogenic,ŌĆÖ and ŌĆśpathogenicŌĆÖ [86]. More expertise is required to interpret the results of various genetic evaluations. Proper genetic sampling, sample processing, adequate sequencing, and precise interpretation, correlated with the clinical situation, constitute the core competence in the field of cardiovascular genetics.
There are few data about whether an asymptomatic carrier of a pathogenic variant needs an ICD. Therefore, current guidelines still do not recommend the genotype-guided ICD as a primary prevention for SCD. However, sophisticated stratification for SCD risk using functional data, such as patch-clamp electrophysiology testing in induced pluripotent stem cells, will be implemented in cardiovascular genetics clinics as part of cascade screening for family members.
CONCLUSIONS
IAs are a group of primary electrical disorders that potentially predispose young individuals, with no evidence of structural heart disease, to SCD. Recent studies have suggested that a substantial proportion of SCD is due to IAs in East Asian people. BrS, LQTS, and IVF constitute the majority of IAs. Patients should be educated to avoid triggers, and an ICD is recommended for secondary prevention. Although each disease may have pathognomonic ECG findings, systemic evaluation is crucial for the diagnosis, and the role of genetic testing is expanding. Advances in genomic sequencing technology will facilitate identification of the underlying genetic pathophysiology of IA syndrome, and will propel physicians into the era of precision medicine in the field of cardiology.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





