


INTRODUCTION
Myelodysplastic syndromes (MDSs) are a group of clonal hematopoietic disorders marked by ineffective hematopoiesis, peripheral cytopenias, and an increased risk of transformation to acute myeloid leukemia (AML). MDS has heterogeneous clinical manifestations and treatment outcomes. The treatment for MDS is based on prognostic factors that predict survival and the characteristics of progression to AML.
MDS is classified based on pathology or prognosis scoring, which subsequently determines the clinical treatment strategy. Intermediate-2 and High group patients according to the International Prognostic Scoring System (IPSS) belong to the high-risk MDS category [1]. The very high, high, or intermediate groups in the Revised International Prognostic Scoring System (R-IPSS) of classification correspond to high-risk MDS [2]. According to the World Health Organization system of classification, high-risk MDS patients are classified into the histological subtypes of refractory anemia with excess blasts-1 (RAEB-1) and RAEB-2 with a median overall survival (OS) of less than 2 years [1-4].
Allogeneic stem cell transplantation is the preferred curative treatment for high-risk MDS patients. However, epigenetic therapy as part of the treatment regime has benefitted MDS patients immensely. Hypomethylating agents improve the quality of life and survival by decreasing the transfusion requirements and inhibiting leukemic transformation. In the United States and Europe, hypomethylating agents have been approved to treat intermediate-2 and high-risk MDS patients [5-10].
Cancer and Leukemia Group B 9221 [5,6] and AZA-001 [7] were two randomized multicenter trials that studied the clinical efficacy of azacitidine in higher-risk MDS patients. Multiple studies showed that decitabine, a DNA methyltransferase inhibitor, showed overall response rates of 30% to 60% in MDS patients from Western countries [8-11]. Patients treated with a dose of 15 mg/ m2 decitabine every 8 hours for 3 days showed a higher overall response rate (ORR, 17%) and complete response in 9% MDS patients [9]. Moreover, 5-day intravenous injections of 20 mg/m2 decitabine showed a 39% complete response rate [10].
Unlike conventional chemotherapy, hypomethylating agents require several cycles to achieve effective results. Pretreatment risk stratification is based on IPSS, R-IPSS, and WHO classification-based prognostic scoring system (WPSS) staging of MDS patients. Patients with high baseline lactate dehydrogenase levels are associated with poor OS when treated with azacitidine [12]. Moreover, previous low-dose cytarabine, > 15% bone marrow (BM) blast counts, poor performance, and transfusion of > 4 units of red blood cells every 8 weeks are factors that predict a low response rate to 5-azacitidine [8].
Biomarkers that predict drug response are required to identify patients that would clinically benefit from treatment with hypomethylating agents. Ten-eleven-translocation 2 (TET2) and DNA methyltransferase gene 3A (DNMT3A) genes are two candidate predictive biomarkers that can be considered for therapy with hypomethylating agents. TET2 is a tumor suppressor gene at chromosome 4q24, which is associated with loss of heterozygosity and passive DNA methylation due to oxidation of 5-methylcytosine to 5-hydroxylmethylcytosine in many hematological malignancies such as myeloproliferative neoplasm (MPN) (10%), MDS (20%), chronic myelomonocytic leukemia (CMML) (30%), and secondary AML [13]. DNMT3A encodes an enzyme that transfers methyl groups to specific CpG dinucleotides in the DNA [14]. CpG methylation is an epigenetic modification that is important for embryonic development as well as regulating gene expression for cellular function.
Papaemmanuil et al. [15] analyzed 111 genes in 738 MDS patients and showed that 78% of the patients had genetic mutations in about 43 genes. Genetic alterations or mutations have been identified in methylation pathway genes such as TET2, DNMT3A, and IDH/IDH2 [13,14,16-19]. Mutations in methylation machinery genes were detected in 10% to 30% of MDS patients with splicing factor SF3B1 mutated in 24% of the patients in the cohort followed by TET2 (22%) and SRSF2 (14%) [15]. Haferlach et al. [20] examined 104 mutational genes in 944 MDS patients and found 47 significantly mutated genes including TET2, SF3B1, ASXL1, DNMT3A, and RUNX1, which were mutated in > 10% of patients. In patients with higher-risk MDS with low blast counts, TET2 status was associated with response to azacitidine, independent of the karyotype [21]. In another study, mutations in TET2, DNMT3A, and IDH1/IDH2 genes were found in 18%, 9%, and 8% of the MDS patients [17].
TET2 and/or DNMT3A mutations were associated with better ORR and favorable progression-free survival (PFS) upon treatment with DNMT inhibitors in MDS patients; ASXL1 and SF3B1 were associated with better OS [17]. However, these results were limited by the low incidence of response to hypomethylating agents, heterogeneous disease entities (MDS, MPN, and secondary AML), and heterogeneous treatment for the low-risk and intermediate-1 risk groups. Therefore, in this study, we analyzed if methylation machinery gene mutations influence the response of MDS patients to decitabine, a demethylating agent.
METHODS
Study subjects and data collection
We recruited 104 patients diagnosed with de novo MDS between January 2007 and December 2011. Pretreated BM samples with adequate genomic DNA were available for these patients. Thirty-four patients were excluded, because their genomic DNA was not adequate for targeted sequencing. Data regarding clinicopathological parameters and treatment outcomes such as patient demographics, Eastern Cooperative Oncology Group performance status, laboratory data, disease stage, OS, and treatment response were obtained retrospectively from patient records.
We analyzed treatment outcomes of MDS patients treated with 20 mg/m2 administered intravenously decitabine daily for 5 consecutive days, repeated every 4 weeks according to the 2006 international working group criteria [22]. This study was approved by the Samsung Medical Center Institutional Review Board (IRB 2012-01-081). The requirement for informed consent for the use of archived tissue samples and retrospective clinical data was waived, and the data was de-identified.
Statistical analysis
Data regarding clinicopathological parameters were analyzed using chi-square and Fisher’s exact tests (categorical variables). Kaplan-Meier survival analysis was performed using the log-rank test. The independent prognostic potential of all clinicopathological parameters was determined via multivariate analysis in accordance with stepwise Cox proportional hazards regression model. Statistical analyzes were performed using the SPSS 19.0 software (IBM Co., Armonk, NY, USA), and results were considered statistically significant at p ≤ 0.05.
We analyzed prognostic factors associated with OS and PFS. OS was defined as the time from the date of the first decitabine treatment to the date of death or the date of the last follow-up visit. Complete remission (CR) was defined as recovery of morphologically normal BM and blood counts (neutrophils ≥ 1,000/µL, hemoglobin [Hb] 11 g/dL and platelets ≥ 100,000/µL) and absence of circulating leukemic blasts or extramedullary leukemia. Relapse was defined by the occurrence of ≥ 5% BM blasts, circulating leukemic blasts, or development of extramedullary leukemia. Progression was defined as return to pretreatment BM blast percentage, decrement of ≥ 50% from maximum remission/response levels in granulocytes or platelets, or reduction in Hb concentration by 1.5 g/dL or transfusion dependence. PFS was defined as the time from the date of decitabine treatment to the date of relapse, death, or last follow-up visit.
Gene mutation analysis
Genomic DNA was extracted from B) aspirate samples using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer’s protocol. We performed direct sequencing of polymerase chain reaction products from patient samples for TET2 (targeted exon 3–11) and DNMT3A (targeted exon 7–23) genes using the BigDye Terminator Cycle Sequencing Ready Reaction Kit on ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) and analyzed the mutations using the Sequencer program (Gene Codes Corp., Ann Arbor, MI, USA). Sequence variants were identified using the GenBank reference sequences NM_017928.4 for TET2 and NM_022552.4 for DNMT3A, respectively. The identified variants were interpreted and classified as either germline or somatic according to the population database (dbSNP, National Center for Biotechnology Information, Bethesda, MD, USA) and somatic mutation database (Catalogue of Somatic Mutations in Cancer). Variants with a frequency of 1% or more were considered to be polymorphisms.
RESULTS
Patient characteristics
Seventy of 104 patients in the study cohort received decitabine treatment for MDS between January 2007 and September 2011 (Fig. 1). Supplementary Fig. 1 shows the results of the direct sequencing analysis of TET2 and DNMT3A mutations. Demographics of the 70 patients are listed in Table 1. The median age of these patients was 67.6 years (range, 18 to 83) with male: female ratio of 3.36:1. We classified the patients into low (five patients, 7.1%), intermediate-1 (32 patients, 45.7%), intermediate-2 (25 patients, 35.7%), and high (8 patients, 11.4%) risk categories according to IPSS. Based on the mutations in the methylation machinery genes, the patients were categorized into wild type (n = 58) and methylation machinery genes-mutated group (n = 12). There were no differences in the baseline characteristics of patients in the mutant or wild type groups.
After failure of decitabine treatment, all patients received supportive care. No one received stem cell transplantation as sequential treatment after decitabine treatment.
Treatment response
The 70 MDS patients received 290 cycles of decitabine treatment. The median follow-up time was 18.3 months (range, 1.0 to 67.2). The median number of decitabine treatment cycles was 4 (range, 1 to 25 cycles). The median number of cycles for any response was 2 (range, 1 to 12 cycles), whereas the median time for best response was 3.3 months. As shown in Table 2, 37 out of 70 MDS patients (52.9%) that received decitabine showed CR, partial response (PR), marrow complete response (mCR), or hematologic improvement (HI). Among these 37 patients, 22 (59.5%) showed response within two cycles of decitabine treatment, whereas 15 (40.5 %) showed response after two cycles of decitabine treatment. The mutation profiles and treatment responses of the MDS patients are shown in Table 3.
Treatment response, survival, and predictive factors
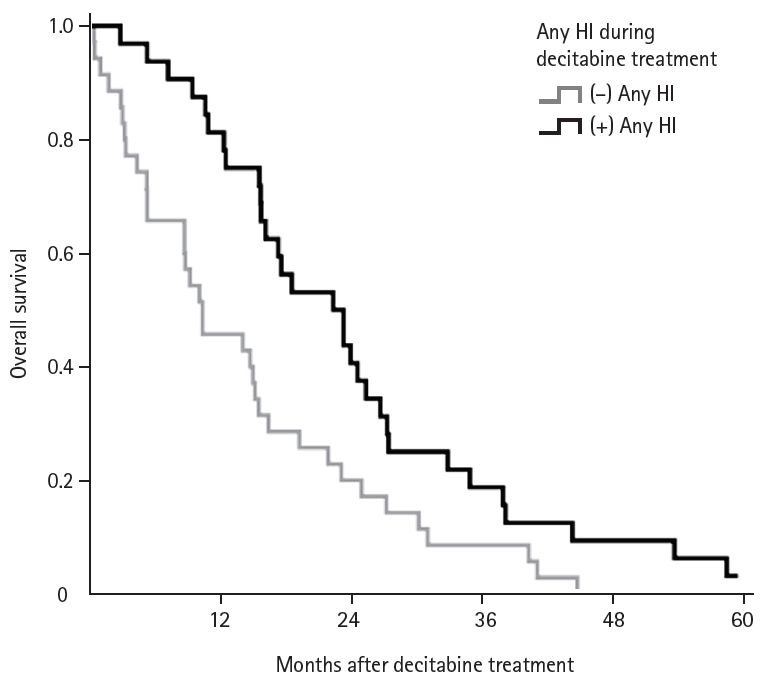
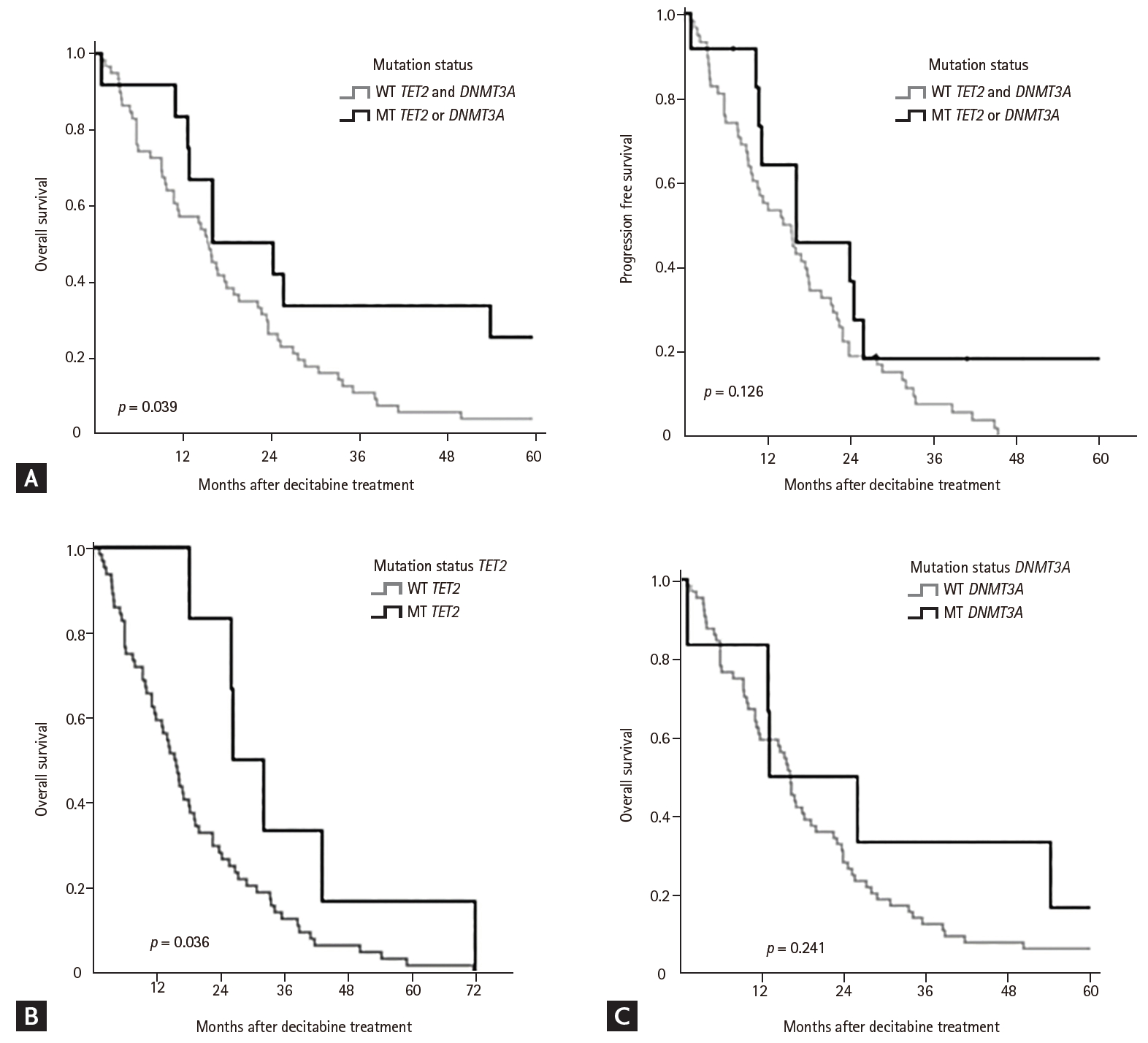
The median OS was 16.1 months (range, 0.9 to 60.8), whereas the median PFS was 15.8 months (range, 0.8to 55.3). Univariate analysis showed that IPSS, WHO classification, and mutation status were factors that predicted ORR (CR + PR + m-CR + HI) (Table 4). The median number of cycles of decitabine treatment was 4 for the wild type group and 7 for the mutant group. Patients with HI, CR, or PR response continued treatment with decitabine, resulting in different treatment cycles between the two groups. In the mutant group, there were more patients who had HI, CR, or PR response (83.3% vs. 47.6%); therefore number of decitabine treatment cycles was different between two groups. Moreover, in total population, the patients with any HI during decitabine treatment showed higher median OS than the patients without HI during or after decitabine treatment (22.9 vs. 10.9 months, p = 0.006) (Fig. 2). Univariate analysis showed that intermediate and high-risk scores according to IPSS and R-IPSS were negative prognostic indicators for OS and PFS (Table 4). Moreover, mutation status of methylation machinery genes was associated with ORR and OS for decitabine treatment, but did not affect PFS. Mutation status of methylation machinery genes was associated with OS (Fig. 3A). The patients with TET2 mutation had better OS than those with wild type TET2 (Fig. 3B). There was difference in survival between wild type DNMT3A and DNMT3A mutant groups (Fig. 3C). As shown in Table 5, multivariate analysis showed that IPSS risk and mutation status were associated with OS.
Treatment outcomes in patients with bone marrow blasts > 5% (n = 38)
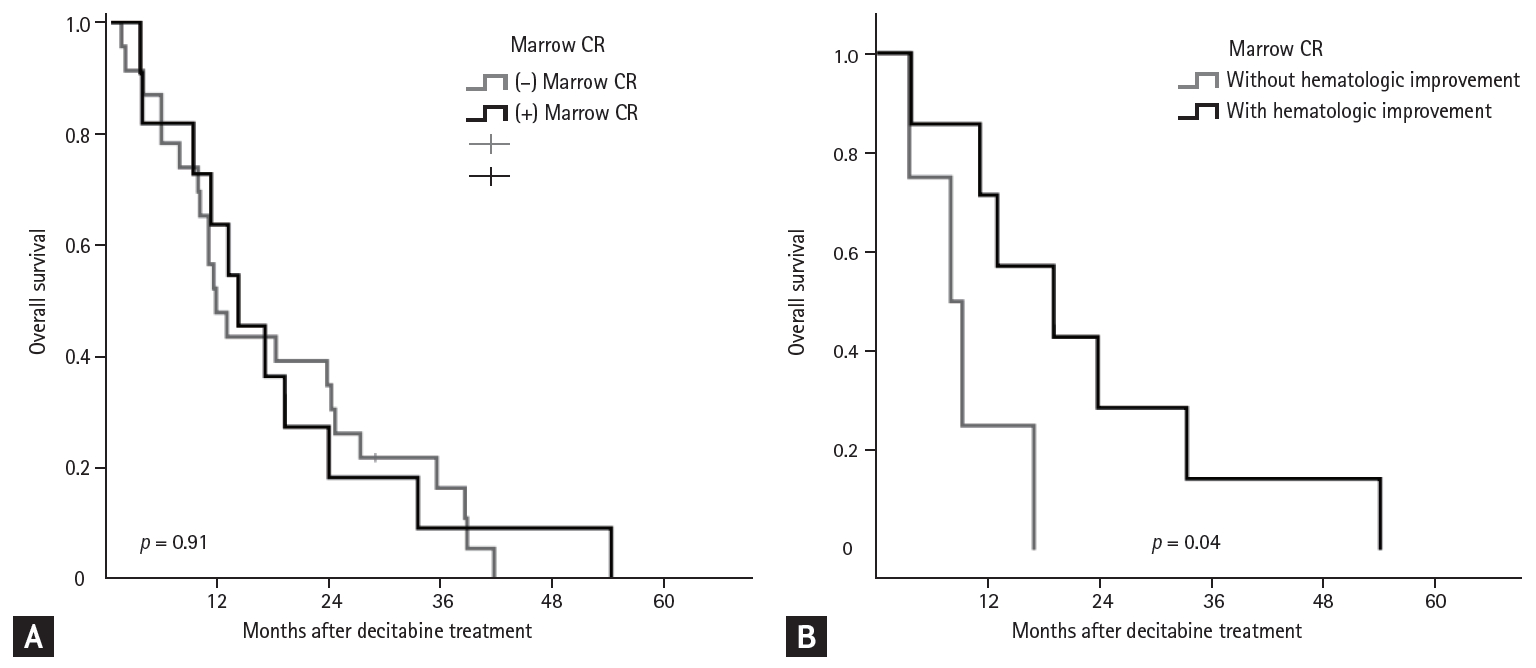
Fig. 4A shows the relationship between m-CR and OS in patients with BM blasts > 5%. The OS was similar in patients with or without m-CR (p = 0.91). However, in subgroup with m-CR, patients with HI showed better OS than patients without HI (19.1 months vs. 8.0 months, p = 0.04) (Table 6 and Fig. 4B).
DISCUSSION
Cancer treatment strategies are constantly changing with the discovery of novel molecular mutations. In this study, we demonstrate that integrating methylation machinery gene mutations, such as TET2, helps obtain improved prognostic information in MDS patients that receive decitabine treatment, thereby providing a basis for personalized clinical treatment strategy.
The genetic basis of MDS is heterogeneous, and a combination of many different somatic mutations results in a diverse pathological manifestation and prognosis. Cytogenetic, genetic, and epigenetic aberrations are all involved in the pathophysiology of MDS. In MDS patients, recurrent mutations have been reported in more than 40 genes that regulate DNA methylation, post-translational chromatin modification, transcriptional regulation, RNA spliceosome machinery, cohesion complexes, and signal transduction. Aberrant DNA methylation is a critical aspect of the altered epigenetic regulation in MDS [13].
Nearly 40% to 60% of high-risk MDS patients respond to treatment with hypomethylating agents, showing a median response duration of 9 to 15 months. Patients that do not respond to hypomethylating agents are associated with poor outcomes and have a median survival rate of less than 5 months [23,24]. Gene mutations have been implicated in the pathogenesis and prognosis of MDS. The significance of TET2 mutations in MDS prognosis is still under debate. Large cohort studies show that TET2 mutations do not affect survival [25,26]. Kosmider et al. [27] demonstrated that TET2 mutations are negatively associated with the prognosis of CMML patients. In contrast, TET2 mutations predict a more favorable response to hypomethylating agents in high-risk patients [28-30]. DNMT3A mutations predict a positive response to hypomethylating agents [17]. In our study, patients with TET2 or DNMT3A mutations showed a higher response rate to decitabine, but this did not translate into a survival benefit. The median number of cycles of decitabine treatment was four in the wild type group and seven in the mutation group. Disease progression shortened the treatment duration in the wild type group. Our findings suggest that screening for specific genetic mutations can identify the subset of patients that respond to hypomethylating therapy. The mechanism by which TET2 mutations influence response to hypomethylating agents is not clear. Patients with TET2 mutations and animal models of Tet2 deletion show altered methylation patterns. However, the extent of DNA methylation induced by pretreatment is not associated with the response to hypomethylating agents.
There are potential limitations to our findings. First, our study is retrospective. Second, we assessed only a small number of MDS cases (n = 70) and also included CMML cases. In this study, the incidence of TET2 or DMNT3 mutation was lower than that in other studies. There are several potential reasons for this, such as race differences, small scale of the study, or problems with sample quality. We analyzed TET2 or the mutation for OS statistics due to a small sample size. In future, we would need a large-scale study for each mutation. Also, this study did not validate the findings using a germline control. Despite these limitations, we based our findings on the available clinical and hematological data, which were adjusted for all potential confounding factors. Since the previous study included a heterogenous population in terms of treatment strategy and patient demographics, we assessed the efficacy of decitabine in a homogenous group of patients to identify predictors for response to decitabine therapy.
The cohort of patients in the current study was homogenous in terms of treatment and disease status and showed comparable responses to hypomethylating agents. Furthermore, BM samples for mutation screening were collected before treatment. The somatic mutations in the tested genes have been well established from large-scale genomic studies, thereby allowing confident predictions to be made. In the future, prospective studies, including studies with large cohorts of patients receiving decitabine are warranted to confirm our findings.
In conclusion, our study shows that mutations in the methylation machinery gene, TET2, are predictive markers for evaluating the response of MDS patients to hypomethylating agents.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement 1
Supplement 1 Print
Print





