


INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is the most common progressive disorder in kidneys characterized by the development of large fluid-filled kidney cysts [1]. These cysts compress and destroy the surrounding kidney tissue resulting in a progressive loss of function in the fifth to sixth decades and by the age of 70 leads to end-stage kidney disease (ESKD) in about 50% of patients [2,3]. Although kidney cysts are the main characteristic of the disease, ADPKD also involves other organs and manifests as liver and pancreas cysts, cerebral aneurysms, cardiac valvular abnormalities, and colonic diverticula [4]. Furthermore, although ADPKD shows a typical family pedigree of autosomal dominant genetic diseases, there is a high degree of phenotypic variability among family members even in those carrying the same mutations. This review will focus on recent developments in the diagnosis and treatment of ADPKD. The genetic and clinical characteristics are described in Korean patients with ADPKD and ongoing studies in these patients are discussed.
EPIDEMIOLOGY
ADPKD occurs in all races worldwide. It is difficult to estimate the precise prevalence of ADPKD because the rate of disease progression varies from patient to patient and the severity is also different. Dalgaard [5] estimated the prevalence of ADPKD at birth as 1 in 400 to 1,000. Suwabe et al. [6] reported that the age- and sex-adjusted annual incidence of definite and likely ADPKD diagnosis during 1980 to 2016 was 3.06/100,000 person-years and the point prevalence of definite or likely ADPKD on January 1, 2010, was 68/100,000 population in Olmsted County, Minnesota, USA. The minimum prevalence of ADPKD in 19 European Union countries, however, estimated that the use of renal replacement registry data and published population-based prevalence was 3.29/10,000 [7]. There was no nationwide prevalence survey of ADPKD in Korea, but it has a single public health insurance system, which indirectly shows the prevalence of diseases. According to the Health Care Big Data Hub, the number of patients with ADPKD in Korea was estimated at 5,320 in 2019, based on the International Classification of Diseases, 10th Revision (ICD-10) code Q61.2 (polycystic kidney, autosomal dominant) [8]. In 2019, the population of Korea was about 51 million, therefore, the prevalence of ADPKD patients is estimated to be about 1/10,000. However, Kalatharan et al. [9] reported that the ICD-10 coding algorithm had a sensitivity of 33.7% and a specificity of 86.2% for the identification of ADPKD. This indicates that the ICD-10 code may overlook a large number of ADPKD patients.
While the prevalence of ADPKD in the general population can only be estimated through an indirect method, the proportion of ADPKD patients among ESKD can be found in more detail through several registries. ADPKD is responsible for up to 10% of patients in ESKD [1]. According to data from the European Renal Association-European Dialysis and Transplantation Association (ERA-EDTA) Registry on renal replacement therapy (RRT), the prevalence of RRT due to ADPKD was 91.1 per million population and the percentage of prevalent RRT patients with ADPKD was 9.8% [10]. Data from the ESRD Registry Committee of the Korean Society of Nephrology indicated that cystic kidney disease accounted for 1.6% of patients in 2018 [11].
ETIOLOGY AND PATHOGENESIS
Mutations in two genes, the polycystic kidney disease 1 (PKD1; chromosome 16p13.3) and PKD2 (chromosome 4q21), that are known to cause ADPKD, account for 85% and 15% of cases, respectively [12]. Recently, mutations in two other genes, GANAB and DNAJB11, have been identified in families with mild polycystic kidney disease and variable polycystic liver disease (PLD) [13,14].
PKD1 and PKD2 produce polycystin-1 (PC1) and polycystin-2 (PC2), respectively. PC1 is an integral membrane protein with 11 transmembrane domains, a short cytoplasmic tail, and a large extracellular region and is involved in protein-protein interaction or protein-carbohydrate interactions [15,16]. The C-terminal tail of PC1 interacts with the corresponding region of PC2 to make a PC1-PC2 complex assembled in a 1:3 ratio [17]. PC2 is a calcium-permeable six-transmembrane protein and regulates intracellular Ca2+ concentration [18]. The PC1-PC2 complex localizes to the shaft and basal body of primary cilia and medicates Ca2+ fluxes in repose to mechanical stimulation [19]. ADPKD is understood to be caused by changes in cilia function, Wnt signaling, intracellular calcium homeostasis, 3′-5′-cyclic adenosine monophosphate (cAMP) levels, Ras/mitogen-activated protein kinase signaling, and concentrating capacity by mutations in PKD1 or PKD2. These abnormalities generate de-differentiation, excessive fluid secretion, and proliferation, leading to cyst development [19].
CLINICAL MANIFESTATIONS AND COMPLICATIONS
Kidney manifestations
The main structural change of ADPKD is the enlargement of both kidneys filled with cysts. The kidney cysts vary in size from a few millimeters to tens of centimeters and the size of the kidney also varies from patient to patient. Most ADPKD patients have hypertension before kidney function decline, 35% of children and more than 80% of patients with ESKD have hypertension [4,20]. Decreased kidney function is the most serious kidney complication, leading to ESKD in about 50% of patients by the age of 70 [2,3]. Several risk factors for kidney function decline have been suggested, which will be discussed later. Approximately 60% of patients have back, flank, or abdominal pain related with cyst enlargement [4]. Complications such as cystic hemorrhage, cyst infection, or urinary stones can cause severe pain. Chebib and Torres [21] reported that 20% to 35% of patients have urinary stone, up to 60% of patients have cyst hemorrhage/gross hematuria, and 30% to 50% of patients have urinary tract infection.
Analysis of the initial symptoms and complications of 364 Korean patients in an ADPKD sub-cohort of the Korean Cohort Study for Outcomes in Patients with Chronic Kidney Disease (KNOW-CKD) [22] which enrolled pre-dialysis patients showed that 87.6% of patients had hypertension, 23.4% of patients had hyperuricemia, 29.1% of patients had urinary stones, and 55.9% of patients had hemorrhagic cysts. However, pain (12.9% of patients), hematuria (4.9% of patients), urinary tract infection (2.2% of patients), and kidney cyst infection (4.1% of patients) had a lower prevalence [23]. Lee et al. [24] analyzed clinical characteristics of 166 Korean ADPKD patients which included 29 ESKD patients. They reported that 65% of patients had hypertension, 50% of patients had flank or abdominal pain, 29% of patients had palpable mass, 42% of patients had proteinuria and 18% of patients had hematuria. Fifty-five patients who did computed tomography (CT) scan were showed that 29% of patients had cyst hemorrhage, 27% of patients had urinary stone, 15% of patients had cyst infection, and 14% of patients had pyelonephritis [24]. Hwang et al. [25] reported the clinical characteristics of 34 Korean ADPKD patients who reached ESKD. Among these patients, 85% had hypertension, 69% had gross hematuria, 16% had urinary stones, and 29% had upper urinary tract infection (Table 1).
Extra-renal manifestation
Liver cysts are the most common extra-renal manifestation. They occur in about 80% of patients with ADPKD who are older than 35 years [26]. Severe liver cysts are more common in women and are related to exogenous estrogen use and multiple pregnancies, but the exact mechanism of liver cyst enlargement is not known [26- 28]. However, after the age of 48 years, 58% of females with severe PLD had regression of height-adjusted total liver volume (htTLV), while htTLV continued to increase in males [27].
Intracranial aneurysm (ICAN) is found in about 9% to 12% of patients, with four times higher prevalence than that of the general population [1,29,30]. The prevalence of ICAN is higher in patients with a positive family history of hemorrhagic stroke or ICAN (21.6%) than in those with negative family history of hemorrhagic stroke or ICAN (11.0%; relative risk, 1.968) [30].
Other extra-renal manifestations of ADPKD found frequently, include cysts in other organs (e.g., pancreas, seminal vesicles, and ovaries), valvular diseases, aneurysm of arteries, and colonic diverticulum.
The ADPKD sub-cohort of KNOW-CKD showed that 77.5% of patients had liver cysts, 12.3% of patients had unruptured ICAN, 1.4% of patients had intracranial hemorrhage, and 1.4% of patients had subarachnoid hemorrhage [23]. Lee et al. [24] reported that among 166 patients 58% had liver cysts and 15% had heart valve insufficiency, and 8.4% had cerebral hemorrhage or infarction. Hwang et al. [25] reported that among 34 patients who reached ESKD, 85% had liver cysts and 16% had stroke (Table 1).
ADPKD can present a variety of kidney and extra-renal manifestations and the incidence of these manifestations is variable. The clinical manifestations of Korean ADPKD patients are not different from previously reported patterns in other countries.
DIAGNOSIS
Ultrasonography is used as an initial test in the diagnosis of ADPKD because of its widespread availability, low cost, and safety. Diagnosis using ultrasonography follows unified criteria proposed in 2009. Revised unified criteria can clinically diagnose ADPKD if patients have a family history regardless of genotype and the number of kidney cysts meets the following criteria. The presence of three or more (unilateral or bilateral) kidney cysts is sufficient to establish a diagnosis in individuals aged 15 to 39 years, two or more cysts in each kidney is sufficient for individuals aged 40 to 59 years, and four or more cysts in each kidney is required for individuals age 60 and older [31]. However, it is difficult to detect cysts as small as 2 to 3 mm in diameter with ultrasonography, unless it is performed by an experienced operator. Magnetic resonance image (MRI) or CT with contrast can detect cysts as small as 2 to 3 mm in diameter. According to Pei et al. [32], MRI can diagnose ADPKD with a sensitivity and specificity of 100% using a total of > 10 cysts as a test criterion in subjects who are at risk of ADPKD aged 16 to 29 years. Similar results were obtained in test subjects between the ages of 30 and 40 years old. Less than five cysts can be considered sufficient for disease exclusion [32]. Since the diagnostic criteria using images were developed in patients who had a family history of ADPKD, it is not sufficient to diagnose patients without a family history [31,32]. Therefore, genetic testing may be necessary for patients who do not have a family history but need to confirm a diagnosis of ADPKD. Young individuals under 40 years old who have a family history of ADPKD and want to donate kidneys also require genetic testing.
A standard method for finding the causative genetic mutations in patients with ADPKD has not been established. In particular, sequencing of PKD1 is complicated because two-thirds of the 5′ region (exons 1–33) of PKD1 are duplicated by gene conversion on chromosome 16 within six pseudogenes (PKD1P1–P6). These pseudogenes have a sequence that is 97.7% identical to the original PKD1 gene sequence [33,34]. Previous studies in other countries showed that PKD1 mutations accounted for 69.7% to 85.5% of cases and PKD2 mutations accounted for 14.5% to 30.3% of cases (Table 2) [35-38]. In the coHOrt for genotype-PhenotypE correlation in ADPKD (HOPE-PKD) which is a prospective multicenter Korean ADPKD cohort study, genetic analyses of PKD1/2 using targeted exome sequencing combined with Sanger sequencing in exon 1 of the PKD1 and a multiple ligation probe assay were performed with 749 Korean ADPKD subjects from 524 unrelated families. In 524 families, researchers could only find pathologic variants in 423 (81.4%) families. Among 423 families, 82.3% with pathologic variants had PKD1 mutations and 17.7% had PKD2 mutations [39]. This is a similar result to previous studies, 89.1% of probands or families were found to have pathologic variants in the Consortium for Radiologic Imaging Observational Study (CRISP) in the USA [35], 96.1% in a regional cohort in the Brittany region of France (GENKYST) [36], 80.0% in an Italian cohort [37], and 84.5% in the Toronto Genetic Epidemiology Study of PKD (TGESP) in Canada (Table 2) [38]. The proportion of patients without pathologic variants is high in patients without family history. Iliuta et al. [40] analyzed 209 patients enrolled in the TGESP study. They reported that compared to those with a positive family history, in whom 95.3%, the mutation detection rate in patients with de novo disease and indeterminate family history was significantly lower at 55.5%. They concluded that PKD without an apparent family history may be due to de novo disease, missing parental medical records, germline or somatic mosaicism, or mild disease from hypomorphic PKD1 and PKD2 mutations [40].
Differential diagnosis
Diagnosing ADPKD is not difficult if a patient has a typical family history and bilateral kidneys are enlarged by multiple cysts. However, if there is no family history or the number of cysts is not considered by the diagnosis criteria, a differential diagnosis may be necessary from various other inherited cystic kidney diseases (iCKDs). About 100 genes have been discovered as the cause of iCKD. Derangement of these genes mainly results in the dysfunction of primary cilium (ciliopathies) [41]. The iCKD include ADPKD, tuberous sclerosis complex, von Hippel-Lindau disease, autosomal dominant tubulointerstitial kidney disease, and pediatric diseases such as autosomal recessive polycystic kidney disease and nephronophthisis [42].
Although iCKD are genetic disorders, diagnoses of these diseases are based on clinical manifestation rather than genetic diagnosis. However, clinical diagnosis is not always easy because the clinical features of these diseases are often similar. Therefore, genetic testing is important for the accurate diagnosis and treatment of iCKD. Bullich et al. [43] used a gene panel based on targeted next-generation sequencing of 140 genes that caused cystic and glomerular hereditary kidney diseases. They identified disease-causing mutations in 78% of patients with suspected hereditary cystic kidney diseases. The clinical diagnosis was confirmed in 62% of patients and changed in 2% of patients, a molecular diagnosis was made in 14% of patients, and no mutation was identified in 22% of patients [43].
In Korea, a 3-year prospective multicenter study to create a cohort of 800 Korean iCKD probands and establish a customized genetic analysis pipeline that can genotype each iCKD (Clinical Research Information Service: KCT0005580) is in progress. The genetic identification of iCKD will be performed in each case using a targeted gene panel of 89 genes related to cystogenesis or ciliopathy. For those with variants of undetermined significance or no variants found by the gene panel, whole-exome sequencing will take place. A family segregation study will be performed to elucidate the cause of genotype-phenotype discordance, in-family severity discordance, or discordance between kidney and extra-renal manifestations [42].
ASSESSMENT OF THE RAPID PROGRESSION OF KIDNEY DISEASE
ADPKD shows highly variable clinical features, especially in progression to ESKD. Recently, a drug that can delay the progression of kidney disease has been approved, and it has become more important to define patients whose kidney disease progresses rapidly. Risk assessment of kidney progression includes three categories of factors: genetic testing, total kidney volume (TKV), and other clinical characteristics.
Genetic testing
The variable clinical features of ADPKD relate strongly with the gene locus effect. Patients with the PKD1 mutation have a more aggressive course than those with PKD2 mutations. The median age at onset of ESKD was 58 years for PKD1 patients and 79 years for PKD2 patients [36,44,45]. The HOPE-PKD study also showed that PKD1 patients have an earlier onset of ESKD compared to PKD2 patients (64.9 years vs. 72.9 years) [39]. The onset of ESKD in Korean ADPKD patients is older in PKD1 patients and younger in PKD2 patients than previous studies. Further studies are needed to determine whether the causes are genetic or acquired factors.
Furthermore, the PKD1 patients are divided into three groups: nonsense, frameshift, and canonical splice site mutations are grouped as mutations that truncate the protein (PKD1 PT), small in-frame insertion/deletion affecting less than five amino acids are grouped as PKD1 in-frame insertion/deletion (PKD1 IF indel), and nonsynonymous missense or atypical splice site mutations are grouped as PKD1 nontruncating (PKD1 NT) [38]. Table 3 shows the comparison of mutation frequencies between HOPE-PKD and other large cohorts. Korean ADPKD cohort showed similar proportion of PKD1 PT and PKD1 NT [36,38,39,46,47].
In addition, compared with patients with PKD1 PT, patients with PKD IF indel, PKD1 NT, and PKD2 have reduced risks of ESKD (hazard ratio [HR], 0.35, 0.10, and 0.03, respectively) and death (HR, 0.31, 0.20, and 0.18, respectively) [38]. The HOPE-PKD study also showed that when PKD1 PT was used as a reference, the PKD2 had a 0.22 times lower HR in ESKD. However, the ESKD risk of the PKD1 IF indel and PKD1 NT patients were not different from PKD1 PT and the risk of death was no different between the four groups [39]. In the study, the reason why the prognosis of PKD1 NT is different from that of previous studies was not clarified and is expected to be verified through ongoing studies [42].
Total kidney volume
TKV is recognized as a biomarker that can reflect the course of the disease. Since the kidneys continue to enlarge as the cysts grow, kidney function remains relatively stable at the initial stage and falls at later stages of the disease. A CRISP study followed 241 ADPKD patients and demonstrated that initial height-adjusted total kidney volume (htTKV) was the most important factor in predicting a future decline in glomerular filtration rate. They demonstrated that a baseline htTKV of 600 cc/m defined the risk of developing stage 3 chronic kidney disease (CKD) within 8 years most accurately with an area under the curve of 0.84 [48].
Although several methods for measuring TKV have been developed, two methods using stereology (TKVs) and ellipsoid equation (TKVe) are widely used. TKVs and TKVe were strongly correlated [49]. We also confirmed concordance between TKVs and TKVe using image data from 295 HOPE-PKD patients (r2 = 0.941, unpublished data).
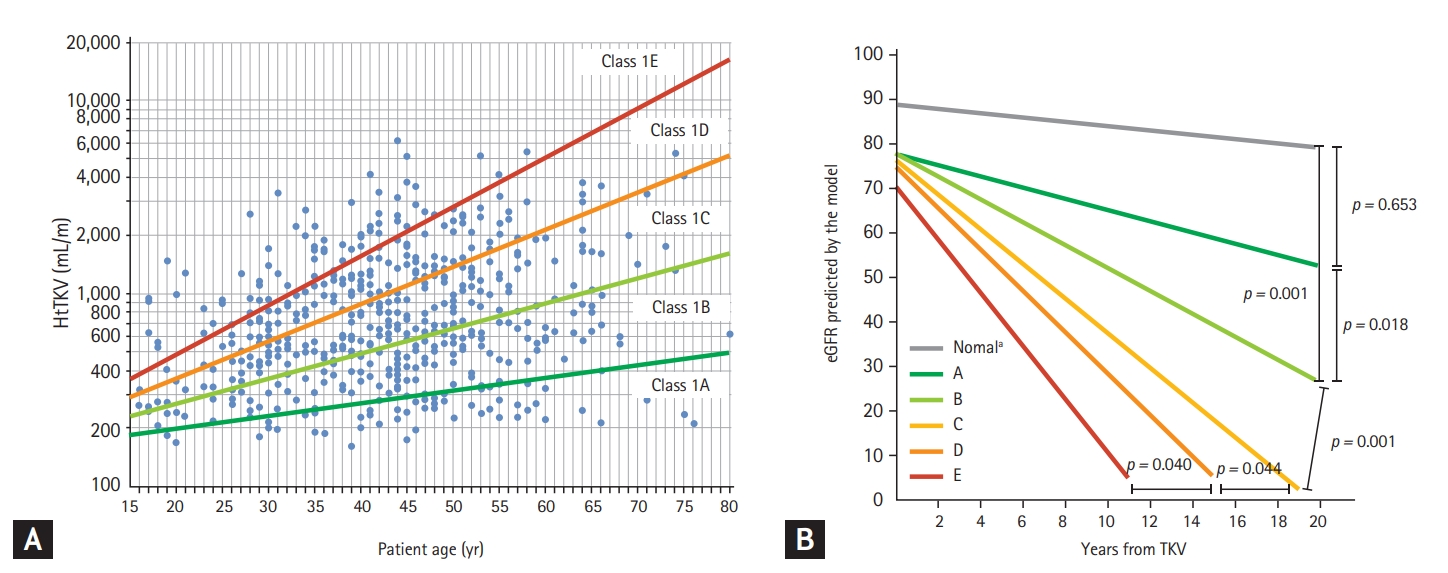
Irazabal et al. [49] further establish this concept in a study of patients from the Mayo Clinic Translational Polycystic Kidney Disease Center (MTPC). This study included a total of 590 patients aged 15 to 80 years. Patients with symmetric distribution of cysts throughout the kidney parenchyma were classified as class 1 and assigned to subclasses on the basis of annual htTKV growth rate estimated from patient age and a theoretical starting htTKV (1A: < 1.5%; 1B: 1.5% to < 3.0%; 1C: 3.0% to < 4.5%; 1D: 4.5% to 6.0%; and 1E: > 6.0%: Mayo Clinic imaging classification) (Fig. 1A). Estimated slopes (mL/min/1.73 m2 per year) of estimated glomerular filtration rate (eGFR) decline by subclass (1A to 1E) are –0.23, –1.33, –2.63, –3.48, and –4.78, respectively, for men and 0.03, –1.13, –2.43, –3.29, and –4.58, respectively, for women (Fig. 1B). The estimated frequency of ESKD at 10 years increased from 1A to 1E (2.4%, 11.0%, 37.8%, 47.1%, and 66.9%, respectively). These were confirmed in the CRISP cohort, a cohort 12 years younger than the MTPC cohort. The estimated frequency of ESKD at 10 years increased from 1C to 1E (2.2%, 14.6%, and 22.3%, respectively) [49]. The study extended the findings of the CRISP with another 5 years of follow-up and showed that the odds ratio of reaching each CKD stage per 100 mL/m increment in htTKV was 1.38 for stage 3, 1.42 for stage 4, and 1.35 for stage 5 or ESKD. This finding further supports the use of htTKV as a strong prognostic and monitoring biomarker in ADPKD [50].
The htTKV and Mayo Imaging classification is measured by the ellipsoid equation using a web-based calculator (http://www.mayo.edu/research/documents/pkd-center-adpkd-classification/doc-20094754).
Other clinical characteristics
The GENCYST study investigators identified four variables that were significantly associated with age at ESKD onset: male, hypertension before 35 years of age, first urologic event before 35 years of age, and PKD mutation type. They developed a new prognostic scoring system (the predicting renal outcomes in ADPKD [PROPKD] score) that accurately predicts kidney outcomes. At 60 years of age, the probabilities of ESKD obtained by Kaplan-Meier analysis were 19.3% in the low-risk group, 60.8% in the intermediate-risk group, and 91.9% in the high-risk group [51].
The ERA-EDTA Working Groups of Inherited Kidney Disorders and European Renal Best Practice have provided algorithms to assess indication for initiation of treatment in ADPKD. They define rapid progression using historical eGFR decline (confirmed eGFR decline ≥ 5 mL/min/1.73 m² in 1 year and/or confirmed eGFR decline ≥ 2.5 mL/min/1.73 m² per year over a period of 5 years or more) and historical kidney growth (an htTKV increase of more than 5% per year using three repeated measurements). They also define likely rapid progression using predicted progression by baseline htTKV indexed for age and/or genotype: htTKV compatible with Mayo class 1C, 1D, 1E, or ultrasound length > 16.5 cm and/or truncating PKD1 mutation + early symptoms (i.e., a PROPKD score > 6). Finally, they define possible rapid progression by a family history of ADPKD that reaches ESKD ≤ 58 years. Working groups have applied these criteria to patients with eGFR ≥ 30 mL/min/1.73 m² and have recommended the use of tolvaptan treatment [52].
Currently, a multinational multicenter retrospective cohort study is in progress to determine the clinical characteristics of rapidly progressing patients with ADPKD from six countries in the Asia-Pacific region: Australia, China, Hong Kong, Korea, Taiwan, and Turkey; using the current recommendation for the identification of rapid progression [53].
TREATMENT
Until tolvaptan was approved as a disease modifying drug, symptomatic therapy was mostly used for the treatment of ADPKD patients. Here, we will discuss the treatments related to kidney symptoms and kidney disease progression of ADPKD. Screening and treatment of extra-renal manifestations will be discussed at other opportunities.
Blood pressure control
Hypertension is the most common kidney symptom in ADPKD and is a major risk factor for kidney failure progression and cardiovascular complications. Blood pressure rises from a relatively young age and is known to be associated with activation of the renin-angiotensin-aldosterone system in the early stages of the disease [54]. The HALT progression of polycystic kidney disease (HALT-PKD) study showed that in early hypertensive ADPKD patients (15 to 49 years of age, eGFR > 60 mL/min/1.73 m2), rigorous blood pressure control (95/60 to 110/75 mmHg) was associated with a slower increase in TKV, no overall change in the eGFR, a greater decline in the left-ventricular-mass index, and greater reduction in urinary albumin excretion compared with standard blood pressure control (120/70 to 130/80 mmHg) [55]. Therefore, it is recommended that blood pressure be adjusted to ≤ 130/80 mmHg, but if the patients are 18 to 50 years old and eGFR > 60 mL/min/1.73 m2 blood pressure control ≤ 110/80 mmHg is recommended. Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers are the first-line therapy for ADPKD [56].
Water intake
In ADPKD, the antidiuretic hormone vasopressin is increased. Vasopressin binds to the vasopressin V2 receptor located in the distal tubule and collecting duct, resulting in increasing intracellular cAMP concentration and activating protein kinase A, which is associated with cyst growth [19]. Therefore, it has been theoretically speculated that drinking a large amount of water may inhibit vasopressin production and the growth of cysts. One expert review recommended that by ingesting water to urinate about 2.5 to 4 L/day and reducing urine osmolarity to ≤ 250 mOsm/kg, it is possible to slow cyst growth by continuously inhibiting vasopressin secretion [57]. Chebib and Torres [56] recommended moderately enhanced hydration spread over 24 hours (during the day, at bedtime, and at night if waking up) to maintain an average urine osmolality of ≤ 280 mOsm/kg.
However, there is no definitive evidence that a large amount of water intake significantly affects changes in TKV and kidney function in patients with ADPKD.
Vasopressin V2 receptor antagonism
The phase 3 Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and its Outcomes (TEMPO 3:4) study was a randomized controlled trial that enrolled 1,445 patients with ADPKD under 50 years of age with TKV > 750 mL and GFR > 60 mL/min (using the Cockcroft-Gault estimation formula). Tolvaptan treatment for 3 years resulted in an increase in kidney volume of 2.8% in the treatment group whereas it was 5.5% in the placebo group, showing an inhibitory effect of about 50%. It also alleviated pain and the rate of kidney function decline [58].
The Replicating Evidence of Preserved Renal Function: An Investigation of Tolvaptan Safety and Efficacy in ADPKD (REPRISE) trial included patients with more advanced ADPKD than the TEMPO 3:4 study. The REPRISE study showed that tolvaptan resulted in a slower kidney function decline than the placebo in patients with later-stage ADPKD over a 1-year period (18 to 55 years of age with an eGFR of 25 to 65 mL/min/1.73 m2 or 56 to 65 years of age with an eGFR of 25 to 44 mL/min/1.73 m2) [59].
There were clinically important increases in liver enzyme concentrations (4.4% in the tolvaptan group vs. 1% in the placebo group) [58]. However, a long-term safety study showed that monthly hepatic monitoring during the first 18 months of tolvaptan exposure and every 3 months thereafter enabled early detection of transaminase elevations and effective intervention [60].
Tolvaptan was approved by the Korean Food and Drug Administration in 2015. Currently, the Evaluating the Safety and Effectiveness in Adult Korean Patients Treated with Tolvaptan for Management of Autosomal Dominant Polycystic Kidney Disease (ESSENTIAL) study is underway to evaluate the safety and effectiveness of tolvaptan in Korean ADPKD patients (NCT03949894).
Cyst infection
Cyst infection and urinary tract infection are common complications in ADPKD patients and; therefore, it is difficult to differentially diagnose kidney cyst and/or hepatic cyst infection. Confirmation involves extracting the cyst fluid from a suspected cyst to identify pathogens or to observe the clinical finding when a fever of 38.5˚C or higher is present for 3 days, accompanied by abdominal pain (usually in a specific area), C-reactive protein elevated by 50 mg/L or more, and cyst bleeding can be excluded [61]. Recently, [18F] fluorodeoxyglucose-labeled white blood cell (WBC) positron emission tomography (PET)/CT has emerged as effective imaging test for diagnosing cyst infections. In a study of 19 Korean patients with ADPKD suspected cyst infection, WBC-PET/CT detected 64% of cyst infection cases, whereas conventional imaging detected 50%. WBC-PET/CT showed false-positive results in two of five cases with no cyst infection [62].
The fluoroquinolone series, which has good cyst penetrability, is used as the primary empirical antibiotic for treatment, and changes according to the culture result and treatment response. It is usually recommended to maintain antibiotics for 4 to 6 weeks, but further studies are needed on the appropriate duration of antibiotic use, the timing of treatment response evaluation, and the timing of surgical drainage.
Nutrition
In ADPKD patients, many complications may occur due to the mass effect caused by the enlarged kidney or liver. Because Korean patients have a smaller body size than Western patients, there is a possibility that the mass effect is more severe. Korean ADPKD patients who have PLD experience pressure-related organomegaly such as leg edema (20.4%), ascites (16.6%), and hernia (3.6%). The most common moderate to severe pressure-related symptom was abdominal fullness (13.6%), followed by early satiety (10.6%), which results in poor oral intake [63]. A cross-sectional study of Korean ADPKD patients showed that 7.3% of patients were mildly to moderately malnourished (modified subjective global assessment [SGA] score of 4 and 5) and 21.7% of patients were at risk of malnutrition (SGA score of 6). HtTKV and htTKLV was the sole significant predictor of malnutrition after adjusting for other risk factors, including kidney function [64].
Malnutrition is one of the strongest predictors of mortality and morbidity in CKD patients [65-68]. In patients with ADPKD, the decline in kidney function is combined with the increasing size of the kidney and/or liver, and malnutrition is likely to occur. Therefore, a regular evaluation of nutritional status and appropriate treatment are required. It has been reported that bioelectrical impedance analysis as well as SGA can be a useful method when evaluating the nutritional status and impact of abdominal cystic organs in ADPKD patients [69].
As with other CKD patients, individualized strategies are recommended according to each patient’s nutritional status, clinical comorbidities, and disease severity to prevent malnutrition, attenuate the progression of CKD, and decrease mortality [70].
Hyperuricemia
It is well known that uric acid levels rise as kidney function decreases; however, there is controversy as to whether hyperuricemia directly worsens kidney function [71,72]. Although there is no consistent recommendation regarding the treatment of asymptomatic hyperuricemia in CKD patients, most Korean physicians treat asymptomatic hyperuricemia in CKD patients to prevent CKD progression and cerebrocardiovascular complications [73].
A retrospective study reported the association of high serum uric acid levels with early-onset hypertension, large kidney volume, and increased risk of ESKD in ADPKD patients [74], but it failed to show the independent effect of hyperuricemia on kidney progression in Korean ADPKD patients [75]. However, they also demonstrated that the correction of hyperuricemia with uric acid lowering agents may attenuate kidney progression in the early CKD stages [75]. Further prospective studies are needed to verify the impact of hyperuricemia control on disease progression in ADPKD.
Other potential therapies
Several therapeutic targets that may slow kidney cyst progression in ADPKD have been identified and many trials are ongoing to investigate various treatments including mechanistic target of rapamycin inhibitors, somatostatin analogs, reduction therapy against sphingolipids, and tyrosine kinase inhibitors [76]. These studies are expected to provide new and better treatments for patients with ADPKD.
CONCLUSIONS
ADPKD is the most common hereditary kidney disease and is associated with decreased kidney function and ESKD. ADPKD not only causes structural and functional defects in both kidneys but can also accompany various extra-renal complications, therefore, management is needed from the early stages of the disease. ADPKD is known to be caused by defects in the PKD1 and PKD2, which encode proteins PC1 and PC2, respectively. In addition to the establishment of genetic tests and image analysis methods to predict the clinical course of the disease, drug therapy to prevent the progression of the disease has been attempted. It is important for physicians to select high-risk patients with various factors and to treat them at an appropriate time to slow the progression of the disease and prevent complications.
It is expected that the ADPKD sub-cohort of the KNOW-CKD study, the HOPE-PKD study, and the Korean iCKD cohort study will provide data on the genetic and clinical characteristics of Korean ADPKD patients. The ESSENTIAL study will also evaluate the safety and effectiveness of tolvaptan in Korean ADPKD patients. It is expected that through these studies, physicians will be able to better understand the characteristics of Korean ADPKD patients and provide appropriate treatment to these patients.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





