


INTRODUCTION
Kidney transplantation (KT) is still the best treatment choice for end-stage renal disease. However, acute rejection remains to be a common complication in KT and is associated with reduced graft survival [1]. Since 1995, the rates of acute rejection have fallen dramatically [2], partly owing to the increased use of induction therapy using biologics, such as rituximab, basiliximab, bortezomib, and so on, and the development of precise and selective induction agents. Biologics are now widely used in various transplant centers [2,3], either routinely or for high-risk individuals. The wide use of biologics for KT has made way for a new era in novel drug development. Because signal blockade can be achieved more easily using biologics, such as antibodies and soluble receptors, than using chemical drugs, target selection is an effective strategy for the development of novel drugs. Numerous proteins with major roles in KT can be considered as target biologics [4]. Although biologic agents are the key to successful allograft function, their development at a small scale is nearly impossible owing to the high cost of their purification and functional characterization [4ŌĆō6]. Thus, a new approach to the development and evaluation of novel biologics is required.
Biologics, defined as synthetic drugs that are composed of peptide components, have made significant contributions to the treatment of transplant rejection, cancer, and immune disorders that were previously complex to treat [4,7]. Biologics have become extensively used in clinical practice, and it seems obvious that they have opened up a new period in the development of novel drugs. Because signal inhibitors can be more easily achieved using biologics, such as antibodies and soluble receptors, than with chemical drugs, target choice enables the generation of new drugs. Numerous proteins, containing cytokines and chemokines that play major roles in the pathogenesis of various diseases, can be considered as targets for biologics [7,8]. However, it is almost impossible to generate new biologics on a small scale because of their purification and functional characterization are costly. Thus, a new approach to the generation and evaluation of novel biologics, which has a relatively low cost and can be achieved over a short time frame, is needed.
We have previously proposed the minicircle (MC) vector system as an alternative approach for the development and production of biologics. MCs are protein-expressing vectors, in which the bacterial backbone has been removed [9]. As bacterial backbone components are not necessary for gene expression and may induce an immune response in mammalian cells, MCs are considered ideal plasmids for transgene expression in vitro and in vivo [10,11]. MCs are smaller than conventional plasmids, increasing the probability of cell delivery. Moreover, MCs escape gene silencing and show sustained expression because of the unique methylation patterns in these vectors [12]. Owing to these advantages of MCs, we, along with others have used them as tools for transgenic expression in animals. Some studies have delivered MCs encoding natural proteins, including hypoxia-inducible factor-1a, a-1-iduronidase, interferon-╬│ (IFN╬│), interleukin-23 (IL-23), and cytotoxic T lymphocyte-associated antigen-4-Ig (CTLA4Ig) into an animal model to examine the efficiency of MCs [13ŌĆō17]. We also have delivered MCs encoding tocilizumab (anti- soluble interleukin-6 receptor [sIL-6R] antibody) and/or etanercept (tumor necrosis factor receptor 2 [TNFR2]-Fc fusion protein) and evaluated its efficacy in allograft rejection models [18]. Using this method, it is conceptually feasible to induce the production of a protein drug by the host using in vivo transfection of vectors carrying the full sequence of the protein drug.
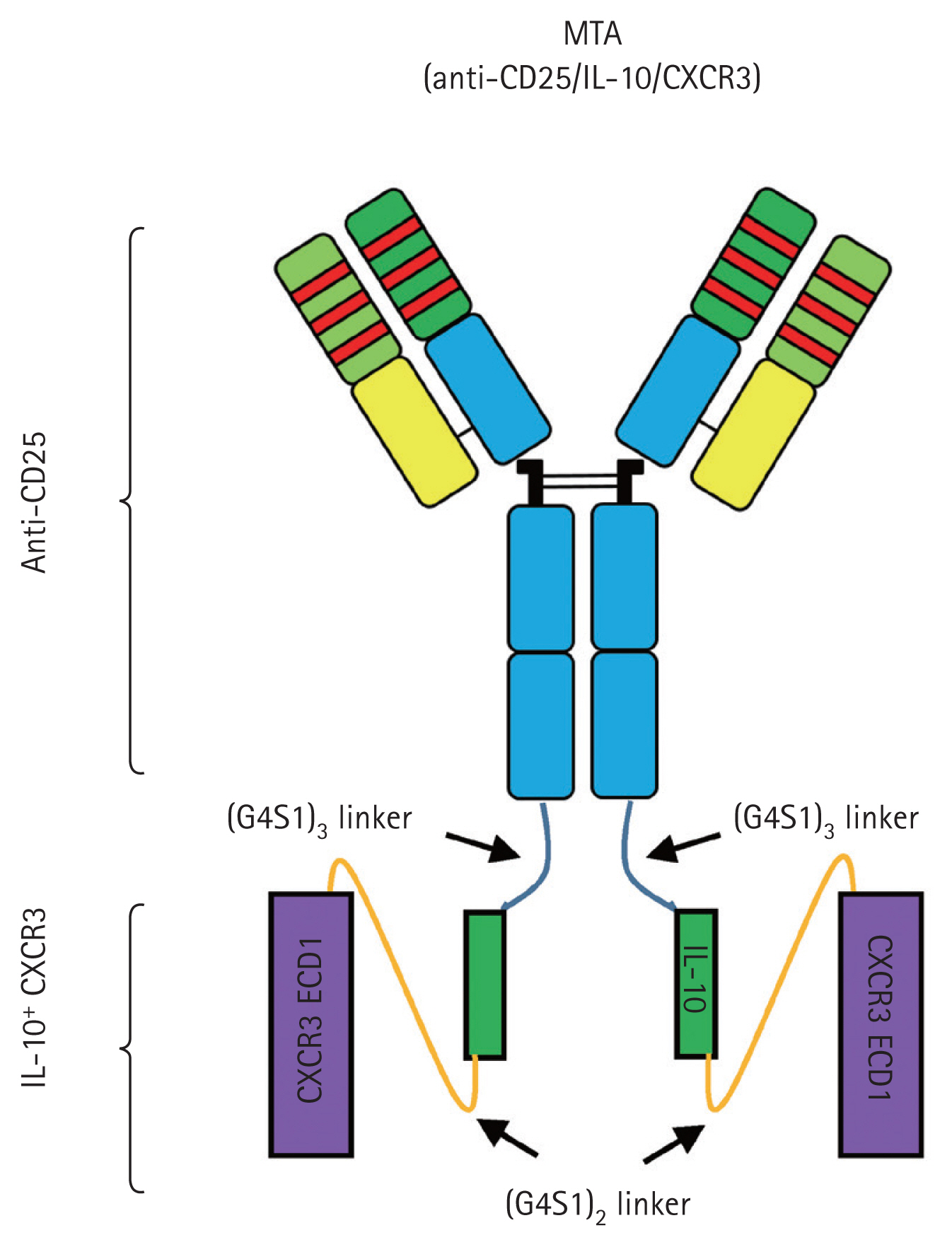
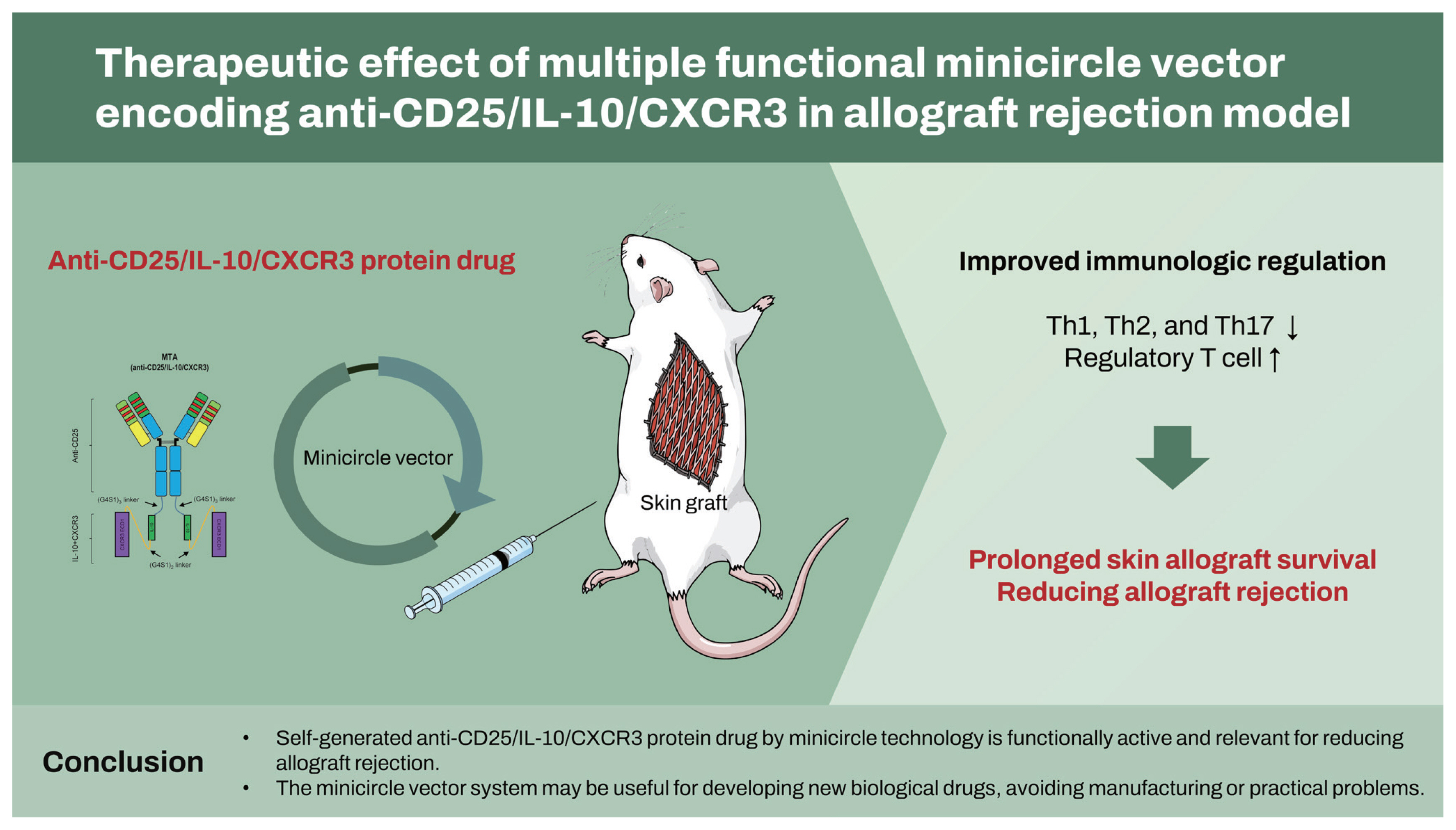
Based on this protein drug delivery platform, we tried to evaluate the therapeutic effect of new synthetic protein on the allograft rejection. Especially in this study, we designed the MCs encoding multiple target-directed agents (MTAs) for the treatment of allograft rejection. We generated MC vectors containing the nucleotide sequences of basiliximab (anti-CD25 antibody), IL-10, and C-X-C motif chemokine receptor 3 (CXCR3), based on the backbone of an MC structure. Treatments using anti-CD25 antibody agents had significantly lower acute rejection rates than induction therapy [19,20]. However, because it also has the same effect on T regulatory (Treg) cells, IL-10 cells conjugated with anti-CD25 antibody compensated for the downregulation of Treg cells [21ŌĆō23], because Treg cells are critical regulators of immune tolerance in transplantation [24,25]. In addition, CXCR3 is a dominant factor directing T cells into the allograft confirmed by highly up-regulated T cells [26]. Based on these findings, we designed the MTA structure by the fusion of the anti-CD25 antibody and IL-10 and CXCR3. In this study, the intravenous injection of the MC encoding MTA, which contained basiliximab, IL-10, and CXCR3 fusion, induced the in vivo production of new synthetic protein drugs. We found that self-produced MTA by the host were functionally active and reduced allograft rejection in a mouse model by using this platform technology for therapeutic protein.
METHODS
Production of minicircles
The MC vectors MC-mock and MC-MTA (anti-CD25/IL-10/ CXCR3) were produced as described previously [27]. It is noteworthy that MTA is a combination of anti-CD25 heavy chain (HC)-IL-10-CXCR3 and anti-CD25 light chain (LC), and anti-CD25 is a combination of MC-anti-CD25HC and LC. To construct the MCs, each sequence (Supplementary Tables 1 and 2) was subcloned into the mock plasmid pMC.EF1-MCS-T2A-RFP (or luciferase [LUC])-SV40 PolyA, which was purchased from System Biosciences (Mountain View, CA, USA). The plasmid contained Nhe1 and BamHI restriction sites, and the cloning outcome was confirmed by double digestion at the two sites. The MCs were produced based on the method previously described by Kay et al. [28]. Briefly, Escherichia coli ZYCY10P3S2T cells were transformed with the protein drug-encoding parent plasmids (PPs). E. coli cells were grown overnight at 37┬░C in Terrific Broth containing 50 ╬╝g/mL of kanamycin and then the LuriaŌĆōBertani broth containing 0.02% l-(+)-arabinose was added to the E. coli cultures. The mixture was incubated at 30┬░C for 5 hours, and MC DNA was isolated using Nucleobond Xtra Purification Kits (Macherey-Nagel, Duren, Germany), according to the manufacturerŌĆÖs instructions.
Cell culture and MC transfection
HEK293T cells were cultured in DulbeccoŌĆÖs modified EagleŌĆÖs medium (DMEM; Gibco, Gaithersburg, MD, USA), supplemented with 7.5% fetal bovine serum (Gibco), 100-U/mL penicillin, and 100-╬╝g/mL streptomycin. HEK293T cells were transfected with the protein drug-encoding MC vectors using the lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturerŌĆÖs instructions. Briefly, MC-anti-CD25HC-IL-10-CXCR3 and MC-anti-CD25LC were co-transfected into cells to produce MC-MTA. MC-mock was transfected alone as the control groups. Cells were transfected with lipofectamine alone in the negative control group. The expression of red fluorescence protein (RFP) in the cells was assessed using fluorescence microscopy at 48 hours post-transfection. The transfected cell culture medium was collected for enzyme-linked immunosorbent assay (ELISA) to quantify the levels of the protein drug. The media were replaced 24 hours before collection, and conditioned media were collected at 5 days after transfection.
Ethics statement
All procedures were performed in strict accordance with the recommendations of the ethical guidelines for animal studies. All experimental animal care protocols were approved by the Animal Care and Use Committee of the Catholic University of Korea (CUMC-2018-0130). Animals were sacrificed under xylazine/rompun anesthesia, and every effort was made to minimize animal suffering.
In vivo gene delivery
MCs were delivered 1 day before skin grafting using the hydrodynamic tail-vein injection method [29]. Briefly, 40 ╬╝g of MC-anti-CD25HC-IL-10-CXCR3 and 40 ╬╝g of MC-anti-CD25LC were injected together into the MTA group to induce MC-anti-CD25-IL-10-CXCR3 production in vivo. Additionally, 40 ╬╝g of MC-IL10-anti-CD25HC and 40 ╬╝g of MC-anti-CD25LC were injected together into the dual-target-directed agent encoding anti-CD25/IL-10 (dual target-directed agent [DTA]) group to induce anti-CD25/IL10 production in vivo. Next, 80 ╬╝g of MC-mock was injected into the control MC-mock group. MCs were injected in a volume equivalent to approximately 10% of the body weight of the mice. To increase hydrodynamic pressure, 27-gauge needles were used, and the injection was completed within 5 to 7 seconds. The blood was collected from the mice injected with the indicated MCs for ELISA to quantify protein drug levels. Animals were allowed to live from 0.5 to 40 days after injection. At each time point, injected mice were used for in vivo imaging, and the blood and liver tissues were then collected for ELISA and fluorescence microscopy.
In vivo imaging analysis for MC detection
To prevent the detection of autofluorescence signals from the digestive system, mice were made to fast for 12 hours before in vivo imaging analysis. Hair removal was also performed to prevent the detection of autofluorescence signals from hair. Multispectral images of luminescence were acquired using the Optical in vivo Imaging System-IVIS Lumina XRMS (PerkinElmer, Waltham, MA, USA). Regions of interest were analyzed using the Perkin Elmer imaging software.
ELISA
To analyze the level of anti-CD25, IL-10, and CXCR3 protein expression in transfected cells or injected mice, the culture media from the transfected HEK293T cells or plasma collected from mice injected with MCs were analyzed using ELISA. The levels of IL-10 or CXCR3 were quantified using sandwich ELISA. Briefly, a 96-well microtiter plate was coated with each commercial anti-mouse IL-10 (MAB417-100, R&D Systems, Minneapolis, MN, USA; at a concentration of 1 mg/mL) or anti-mouse CXCR3 (NBP2-41250, Novus Biologicals, Centennial, CO, USA; at a concentration of 1 mg/mL) in coating buffer and was incubated overnight at 4┬░C. The plate was incubated with blocking solution for 1 hours, followed by incubation overnight at 4┬░C with 100 ╬╝L of serially diluted recombinant murine IL-10 (210-10, Pep-proTech, Rocky Hill, NJ, USA; used as a standard) or recombinant mouse CXCR3 (MBS1172599, MyBioSource, San Diego, CA, USA; used as a standard), and the samples of conditioned media or plasma. Anti-mouse IL-10 biotinylated antibodies (BAF417, R&D Systems) or anti-murine CXCR3 biotinylated antibodies (MBS541349, MyBioSource) were applied to the wells and incubated for 2 hours. After incubation with anti-streptavidin-horseradish peroxidase (HRP) (DY998, R&D Systems) for 20 minutes, the TMB substrate solution (DY999, R&D Systems) was applied to each plate for 20 minutes.
Indirect ELISA was performed to determine the levels of anti-CD25. Briefly, a 96-well microtiter plate was coated with recombinant human sCD25 (21-8702-U100, TONBO Biosciences, San Diego, CA, USA) at a concentration of 1 mg/mL in coating buffer and was incubated overnight at 4┬░C. The plate was incubated with blocking solution for 1 hours, followed by incubation with 100 ╬╝L of serially diluted basiliximab (Simulect, Novartis Pharma AG, Basel, Switzerland; used as a standard) and the samples of conditioned media or plasma overnight at 4┬░C. After incubation with anti-human immunoglobulin G (IgG)-HRP (AP112P, Merck Millipore, Billerica, MA, USA) for 2 hours, the TMB substrate solution (DY999, R&D Systems) was applied for 20 minutes. Absorbance was then measured at 405 and 570 nm.
To identify the fusion complex of MTA, a 96-well microtiter plate was coated with recombinant human sCD25 (21-8702-U100, TONBO Biosciences) at a concentration of 1 mg/mL in coating buffer and was incubated overnight at 4┬░C. The plate was incubated with blocking solution for 1 hours. and then anti-human IL-10 biotinylated antibodies (BAF217, R&D Systems) or anti-murine CXCR3 biotinylated antibodies (MBS541349, MyBioSource) were applied to each plate for 2 hours. After incubation with anti-streptavidin-HRP (DY998, R&D Systems) for 20 minutes, the TMB substrate solution (DY999, R&D Systems) was applied for 20 minutes.
Transwell migration assay
The upper chambers have membranes with 8-╬╝m pore size (Corning Incorporated, Kennebunk ME, USA). Before performing migration assay, HEK293T cells were seeded in the upper chambers and transfected MC-MTA. Chemokine (C-X-C motif) ligand (CXCL) 9, 10, and 11 (#250-18, 250-16, 250-29, PeproTech, Cranbury, NJ, USA) were added in the lower chamber after 48 hours. After 24 hours of incubation, migrated cells on the underside of the trans-well membrane were fixed with methanol and stained with 1% toluidine blue. The migrated cells were counted and averaged from six random fields.
Skin allograft rejection in mice
For the experimental skin graft model, the protocol described by McFarland and Rosenberg [30] was employed. Briefly, C57BL/6 mice were used as donors for skin transplantation. Skin from the donor tail (1.0 cm2) was placed on the back of BALB/c recipients. The recipient mice were transplanted with MC-mock injection, MC-DTA, or MC-MTA injection with or without daily administration of tacrolimus (Tac, Astellas Pharma, Ibaraki, Japan; 2 mg/kg/day diluted in distilled water) by gavage for 17 days, starting 3 days prior to transplantation. Graft rejection was defined as the complete destruction or desiccation of the grafted skin on inspection. Graft survival curves were constructed using the KaplanŌĆōMeier method and graft survival was compared between two groups using the log-rank test. The skin grafts were removed at the indicated time points and rinsed in cold saline, immersed in 4% paraformaldehyde for 2 hours at 4┬░C, and embedded in paraffin for histopathologic examination. Tissue sections (5 to 7 ╬╝m) were stained with hematoxylin and eosin (H&E) for the assessment of cell infiltration.
EXPERIMENTAL DESIGN
In the MC-mock group (n = 5), mice were injected with 80 ╬╝g of MC-mock intravenously 1 day before skin transplantation. In the MC-DTA encoding anti-CD25/IL-10 (n = 5), mice were injected with 40 ╬╝g of MC-anti-CD25HC and 40 ╬╝g of MC-anti-CD25LC intravenously 1 day before skin transplantation. In the MC-MTA group (n = 5), mice were injected with 40 ╬╝g of MC-anti-CD25HC-IL-10-CXCR3 and 40 ╬╝g of MC-anti-CD25LC intravenously 1 day before skin transplantation. In the Tac + MC-DTA or MC-MTA groups (n = 5 in each group), mice received 2 mg/kg Tac daily by oral gavage, starting 3 days before transplantation, for 17 days in the MC-DTA and MC-MTA groups.
Immunohistochemistry
Deparaffinized tissue sections were incubated in retrieval solution (pH 6.0), methanolic H2O2, and 0.5% Triton X-100 and were then washed in phosphate-buffered saline. Nonspecific binding sites were blocked in 10% normal donkey serum (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Sections were incubated overnight at 4┬░C with the following antibodies: monoclonal rabbit anti-CD4 (1:100; ab183685, Abcam, Cambridge, UK) and monoclonal rat antibody against mouse IL-10 (1:50; BD559063, BD Biosciences, San Jose, CA, USA). Primary antibody binding was visualized using the following secondary antibodies: Cy3-conjugated goat anti-rabbit antibody (1:1,000; Jackson ImmunoResearch) and Alexa Fluor 488-conjugated goat anti-rat antibody (1:200; ThermoFisher, Waltham, MA, USA). Stained tissues were viewed using a fluorescence microscope (Axio imager M2, Carl Zeiss Co. Ltd., Oberkochen, Germany). Images were converted to the tagged image file format (TIFF), and contrast levels were adjusted using Adobe Photoshop v. 13 (Adobe System, San Jose, CA, USA). Immuno-labeling profiles of skin grafts were evaluated by counting approximately 10 randomly selected areas (22,534.06 ╬╝m2 per field) in each stained tissue section at ├Ś100 magnification using an image analysis software (Celleste version 5.0.2, ThermoFisher).
Flow cytometry
The expression of cytokines and transcription factors was assessed using cell surface and intracellular staining. At day 5 after skin grafting, single-cell suspensions of the spleens of mice were prepared and stained with fluorescently labeled monoclonal antibodies. Splenocytes were stained with various combinations of fluorochrome-conjugated antibodies against CD4 and CD25. Intracellular flow cytometry for cytokines (IFN╬│, IL-4, and IL-17) and the transcription factor forkhead box P3 (Foxp3) was also performed using either Cytofix/Cytoperm (BD Biosciences, San Jose, CA, USA) or fixation/permeabilization buffer (eBioscience, San Jose, CA, USA), according to the manufacturerŌĆÖs instructions. Appropriate isotype controls were always included. All samples were analyzed using a FACS LSRFortessa (BD Biosciences) and FlowJo software (FlowJo LLC, Ashland, OR, USA).
Statistical analysis
Data are expressed as mean ┬▒ standard deviation of at least three independent experiments. Comparisons among groups were performed using one-way analysis of variance with BonferroniŌĆÖs post hoc test using GraphPad Prism version 7.03 for Windows (GraphPad Software, La Jolla, CA, USA). Statistical significance was set at a value of p < 0.05.
RESULTS
Construction and generation of MC encoding MTA
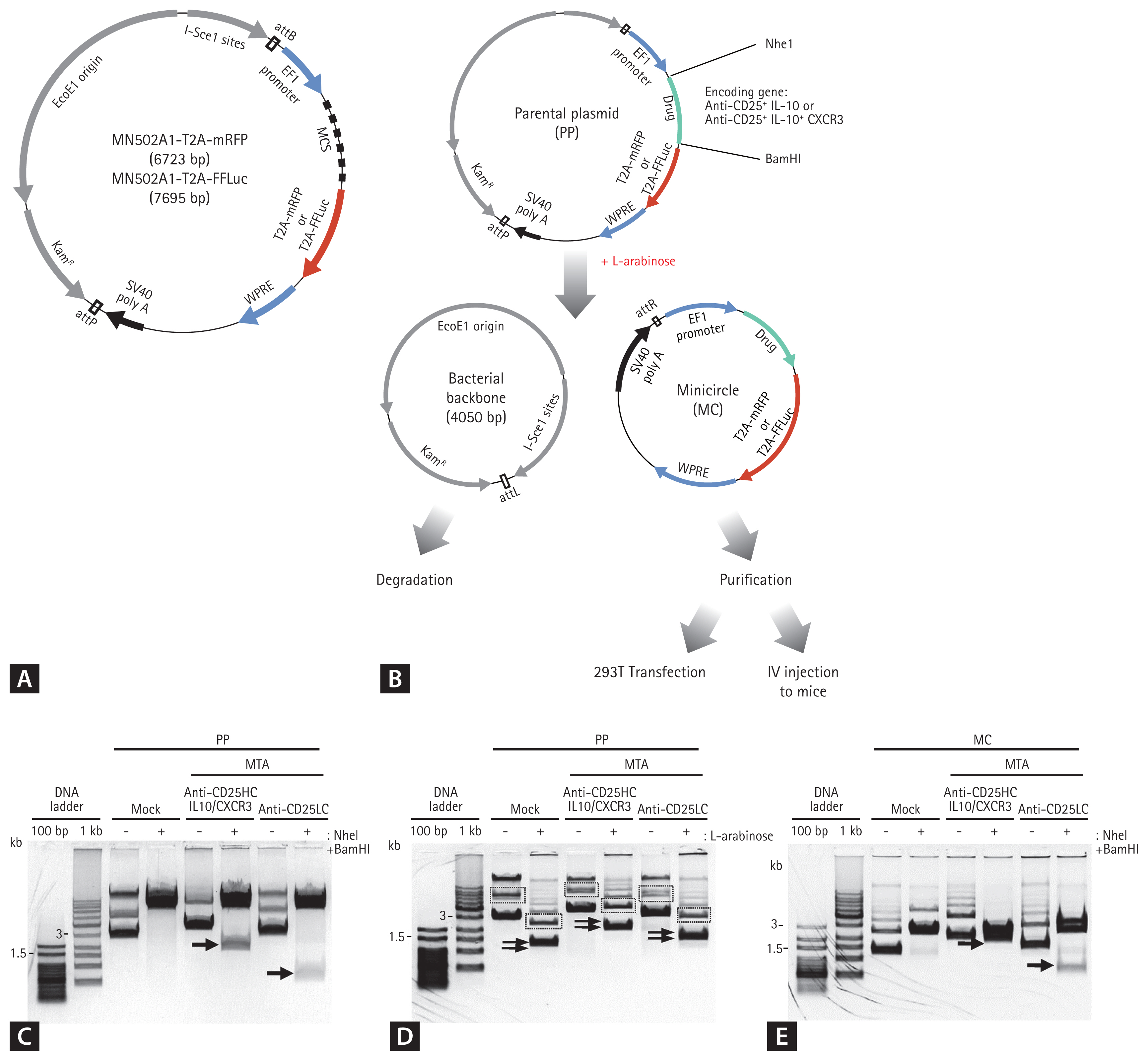
Because the blocking of CD25 and the production of IL-10 in T cells play important roles in preventing allograft rejection, we previously designed a DTA [31]. In this study, we designed the MTA as the CXCR3 sequence conjugated to the anti-CD25 antibody and IL-10 sequence by linkers (Supplementary Tables 1 and 2). To generate the fusion form, the nucleotide sequences of anti-CD25HC attached to the 3ŌĆ▓ end of the sequences encoding the IL-10 and CXCR3 sequence and the anti-CD25LC sequences were subcloned into the PP separately (PP-anti-CD25HC-IL-10-CXCR3 and PP-anti-CD25LC). We predicted that the transfection of both plasmids into cells would lead to the construction of the intact form of the anti-CD25/IL10/CXCR3 fusion protein (Fig. 1). Each insert was subcloned downstream of the EF1 promoter, and RFP or LUC sequences were placed under the control of the EF1 promoter (Fig. 2A).
The two plasmids were transfected into a single cell to generate an intact anti-CD25/IL-10/CXCR3 protein drug. After PPs were treated with l-arabinose, MCs were generated, downsized by phage ╬”C31, and the bacterial backbone was degraded. The ╬”C31 integrase combines the attB and attP sites of the PP vector, resulting in the production of two circular DNAs (the MC plasmid and bacterial backbone) (Fig. 2B). Purified MCs were used for subsequent experiments.
We confirmed that the expressible DNA sequences of each protein drug were subcloned into the PP by digestion with XbaI and BamHI (Fig. 2C). We detected a band corresponding to the size of the insert. We also confirmed that the whole DNA sequences intended for subcloning were contained in the PP by DNA sequencing. Supplementary Table 2 provides the peptide sequences of the protein drugs. We also confirmed that the PPs encoding protein drugs were properly converted to MCs after treatment with l-arabinose (Fig. 2D). After digestion with Nhe1 and BamHI, the DNA fragments of interest were detected in the MCs (Fig. 2E).
Protein drug production in vitro using MC encoding MTA
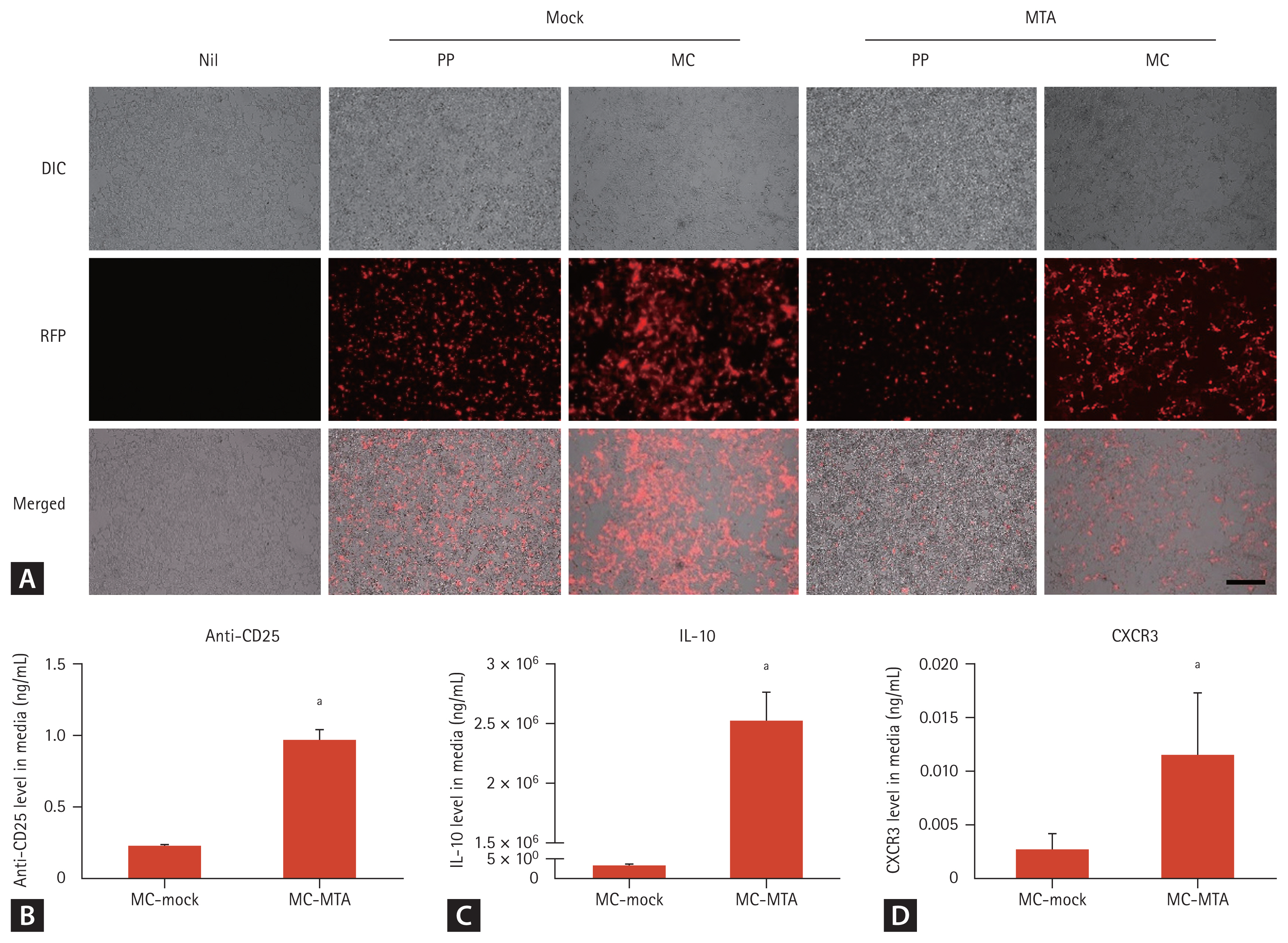
To investigate whether MCs are expressed in vitro, we examined RFP expression after the transfection of PPs and MCs into HEK293T cells. Higher RFP expression was observed by fluorescence microscopy in the MC group than in the PP group (Fig. 3A). The culture media of transfected cells were collected, and the levels of the anti-CD25, IL-10, and CXCR3 proteins were assessed by ELISA using CD25, anti-IL10, and anti-CXCR3-coated plates, respectively. MCs carrying the DNA sequences of the protein drug yielded efficient expression of each drug in the culture media at day 3 (Fig. 3BŌĆō3D).
Protein drug production in vivo using MC encoding MTA
To investigate the persistence of MC expression in vivo, we examined luminescence expression after the hydrodynamic tail vein injection of MC-MTA into mice using in vivo imaging, mainly in the abdomen. Supplementary Figs. 1ŌĆō3 showed no significant differences in body weight and basic functions between the MC-mock and MC-MTA groups (Supplementary Methods).
RFP expression was not observed in mice injected with saline. After MC-MTA injection, RFP levels increased from 1 to 13 days, and the enhanced expression was sustained until at least 40 days, which was the final time point examined (Fig. 4A and 4B). The hydrodynamic delivery method for MC injection temporarily upregulates the permeability of endothelial cells in the capillaries by increasing pressure [32,33]. The liver contains a large number of capillaries and is the most common site for gene delivery using this method [34ŌĆō37]. Therefore, we evaluated RFP-positive cells in the liver tissues of mice injected with MCs. Numerous RFP-expressing cells were detected at 13 days and expression was sustained for 40 days, consistent with the in vivo imaging results (Fig. 4C).
To assess the expression and secretion of anti-CD25/IL-10/CXCR3 derived from MC-MTA, plasma anti-CD25, IL-10, and CXCR3 levels were measured by ELISA on CD25, anti-IL-10, or anti-CXCR3-coated plate using plasma samples at 1, 7, and 30 days after injection with MC-MTA. MCs carrying the DNA sequences of the MC-MTA yielded the efficient expression of each drug at day 7. Anti-CD25/IL10/CXCR3 generation by the MC-MTA was retained until 30 days after MC-MTA treatment (Fig. 5AŌĆō5C). In addition, we compared the concentrations of anti-CD25, IL-10, and CXCR3 in each plasma sample at 3 days after the injection of PP-MTA or MC-MTA and found that the concentrations were higher in the MC-MTA group than in the PP-MTA group, as shown in Fig. 5D and 5F.
Cell migration function by MC encoding MTA in vitro and in vivo
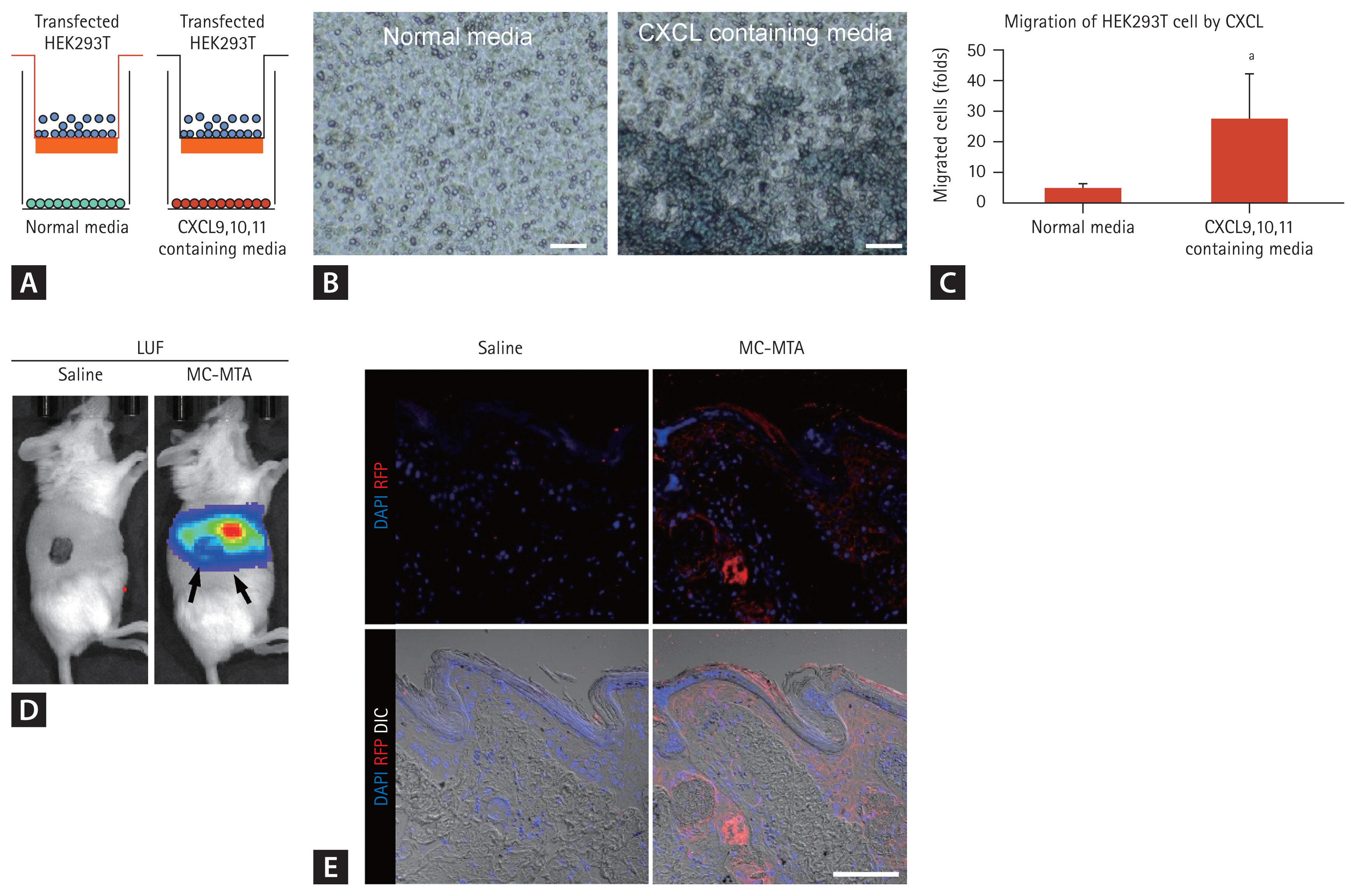
We applied the transwell migration assay in the co-culture system to examine the consequences of increased migrating cells under ligands of CXCR3 e.g., CXCL9,10, and 11 (Fig. 6A). As shown in Fig. 6B and 6C, bottom chamber containing CXCLs showed promoted migration of MC-MTA transfected HEK293T cells into the bottom sides of the insert wells of the upper chamber compared with the normal media group (Fig. 6B and 6C). For in vivo migration of MC-MTA to the allograft, we delivered MC-MTA (tagging LUC or RFP) in the recipient of the skin allograft mouse model. We measured LUC around the transplanted skin allograft and found that LUC was detected in the allograft (Fig. 6D). RFP fluorescence was also stained with anti-RFP antibody compared with the saline group (Fig. 6E).
Therapeutic effect of MC encoding MTA on skin allograft rejection
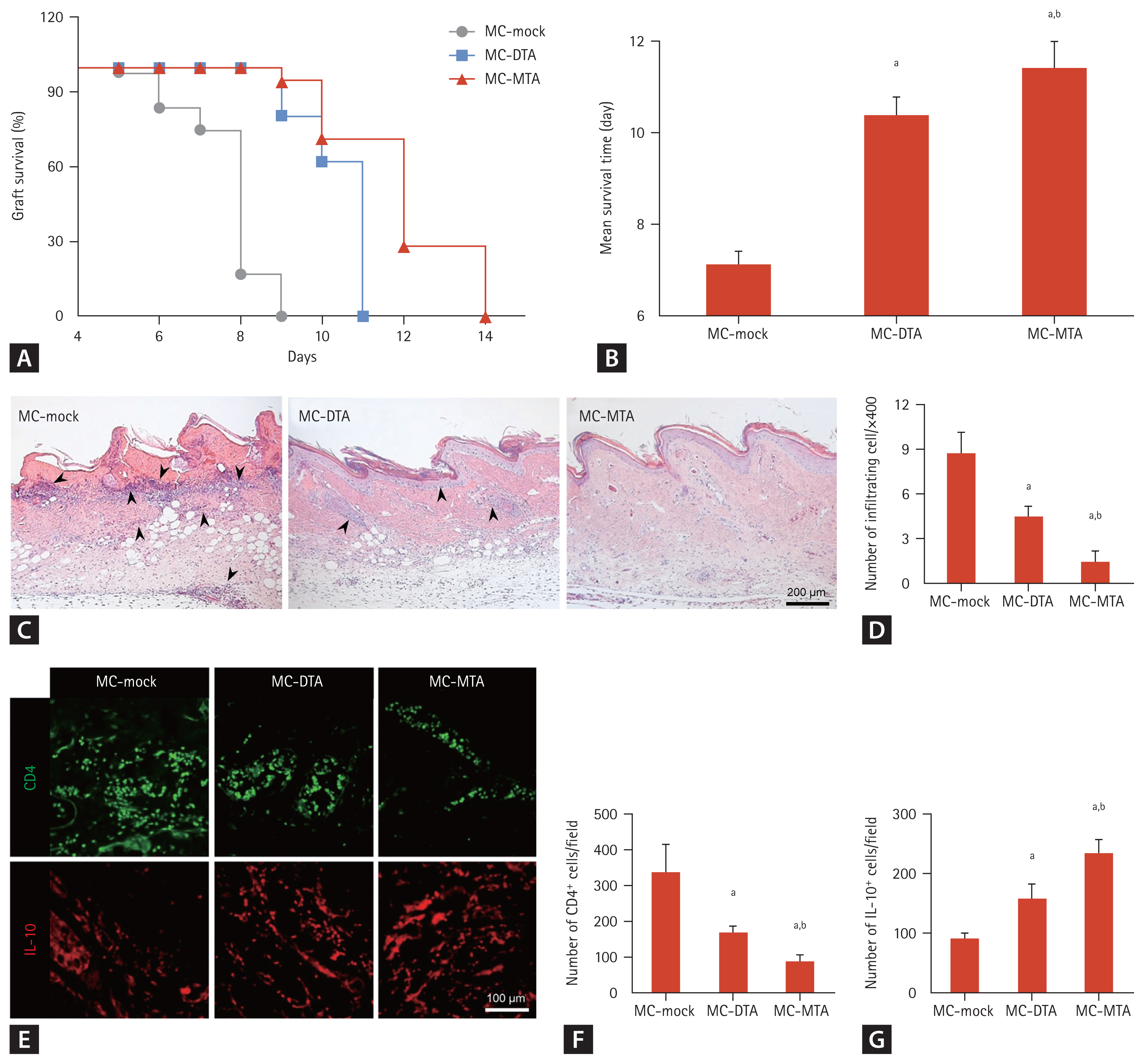
We used a highly immunogenic in vivo experimental skin allograft model to examine the function of the protein drug from the MC encoding anti-CD25/IL-10/CXCR3. One day before receiving a skin allograft, BALB/c recipients were injected with each MC using the hydrodynamic delivery method. Several animals were sacrificed for flow cytometry and H&E staining, and the remainder were visually inspected daily until complete graft rejection. Skin allografts in MC-mock-injected mice had a median survival time (MST) of 7.1 ┬▒ 0.3 days. The treatment of mice with MC-DTA resulted in the significant prolongation of skin allograft survival (10.4 ┬▒ 0.4 days, p < 0.05 vs. MC-mock). Interestingly, MC-MTA-injected mice showed longer MSTs than the other groups (11.4 ┬▒ 0.6 days, p < 0.05 vs. other groups) (Fig. 7A and 7B). We observed marked desmoplasia, hyalinization, and inflammatory cell infiltration in the MC-mock-treated allogeneic control group, but the number of infiltrating cells was reduced in the MC-DTA group (4.5 ┬▒ 0.6 vs. 8.8 ┬▒ 1.4, p < 0.05 vs. MC-mock group), with the greatest reduction in the MC-MTA-injected group (1.5 ┬▒ 0.6, p < 0.05 vs. MC-DTA group) (Fig. 7C and 7D). Fig. 7EŌĆō7G shows immunoreactivity for CD4 or IL-10 and its quantitative analysis in skin allograft tissue section in each group. The number of CD4 positive cells was significantly lower in the MC-DTA group than in the MC-mock group (168 ┬▒ 18/field vs. 339 ┬▒ 76/ field, p < 0.05 vs. MC-mock group). However, the MC-MTA group showed a further reduced number of CD4 positive cells compared with the MC-DTA group (89 ┬▒ 18/field, p < 0.05 vs. MC-DTA group). On the contrary, the MC-MTA group showed a higher number of restored IL-10 cells than the MC-mock and MC-DTA groups (235 ┬▒ 22 vs. 92 ┬▒ 9/ field in the MC-mock group and 159 ┬▒ 25/field in the MC-DTA group, p < 0.05 vs. MC-mock and MC-DTA groups).
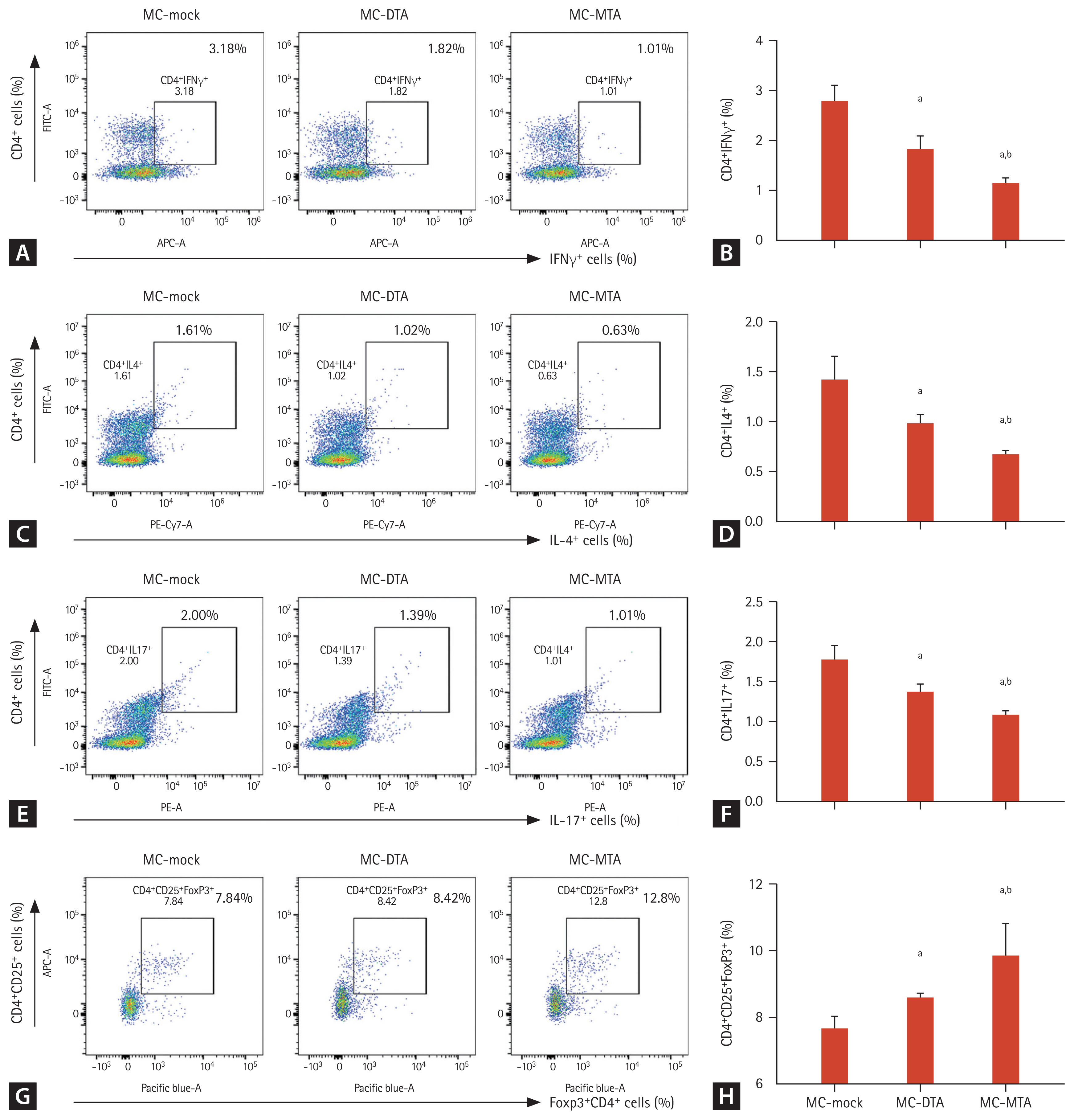
Next, we evaluated whether the prolonged skin allograft survival in the MC-MTA group is associated with the suppression of the pro-inflammatory T cell subset and induction of anti-inflammatory T cells (Treg cells). We performed flow cytometry to evaluate Th1, Th2, Th17, and Treg cells in splenocytes from the experimental animals. As shown in Fig. 8, the numbers of CD4+IFN╬│+, CD4+IL4+, and CD4+IL17+ cells (i.e., Th1, Th2, and Th17 cells, respectively) were significantly lower in the MC-DTA and MC-MTA groups than in the MC-mock group. The number of positive cells was the lowest in the MC-MTA group (Fig. 8AŌĆō8F). The population of CD4+ Treg cells (defined as CD4+CD25+Foxp3+ cells) increased significantly in the MC-DTA group and was the highest in the MC-MTA group than in the MC-mock group (Fig. 8G and 8H).
Combined effect of Tac and MC encoding MTA on skin allograft rejection
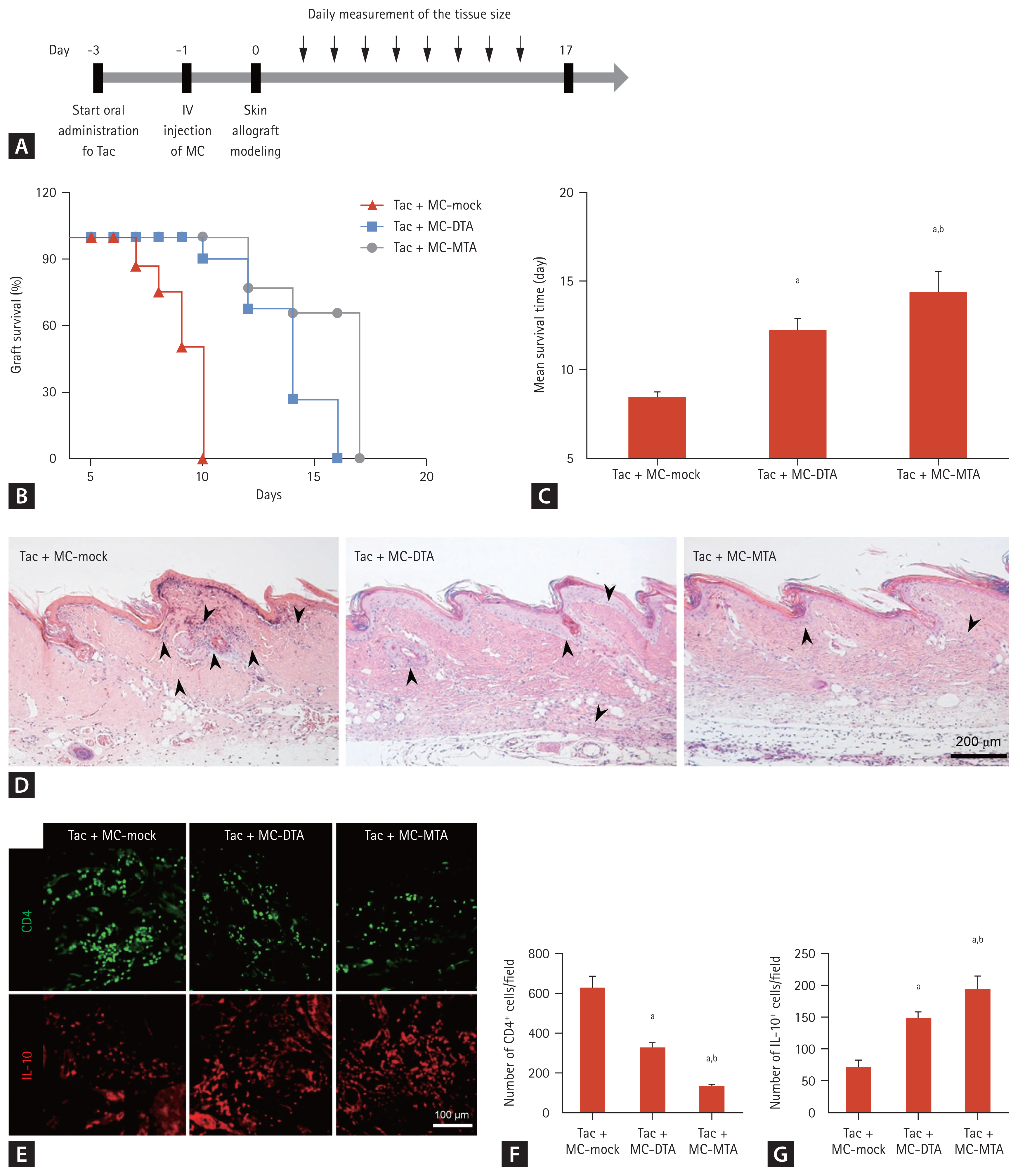
Although Tac is a first-line immunosuppressant in transplantation, it is associated with immunological complications [38ŌĆō40]. Therefore, we compared allograft survival using Tac + MC-mock, Tac + MC-DTA, or Tac + MC-MTA combination. Starting at 3 days before transplantation, recipient BALB/c mice were administered Tac daily, and MC-mock, MC-DTA, or MC-MTA was injected using the hydrodynamic delivery method 1 day before skin allograft modeling. Several animals were sacrificed for flow cytometry and H&E staining, and the remaining animals were visually inspected daily until complete graft rejection (Fig. 9A). Skin allograft survival in the Tac + MC-DTA group was increased compared with that in the Tac + MC-mock group (MST, 12.3 ┬▒ 0.6 days vs. 8.4 ┬▒ 0.3 days, p < 0.05 vs. Tac + MC-mock). The Tac + MC-MTA group had significantly skin allograft survival compared with the Tac + MC-DTA group (MST, 14.4 ┬▒ 1.1 days, p < 0.05 vs. Tac + MC-DTA) (Fig. 9B and 9C). Fig. 9EŌĆō9G show immunoreactivity for CD4 or IL-10 and the results of the quantitative analysis in the skin allograft tissue section in each group. The number of CD4 positive cells was significantly lower in the Tac + MC-DTA group than in the Tac + MC-mock group (324 ┬▒ 30/field vs. 629 ┬▒ 56/field, p < 0.05 vs. Tac + MC-mock group). However, the Tac + MC-MTA group showed a further reduction in the number of CD4 positive cell compared with the Tac + MC-DTA group (132 ┬▒ 6/field, p < 0.05 vs. Tac + MC-DTA group). On the contrary, the Tac + MC-MTA group showed a higher number of restored IL-10 cells than the Tac + MC-mock and Tac + MC-DTA groups (197 ┬▒ 19/field vs. 73 ┬▒ 10/field in the Tac + MC-mock group and 150 ┬▒ 10/field in the Tac + MC-DTA group, p < 0.05 vs. Tac + MC-mock and Tac + MC-DTA groups).
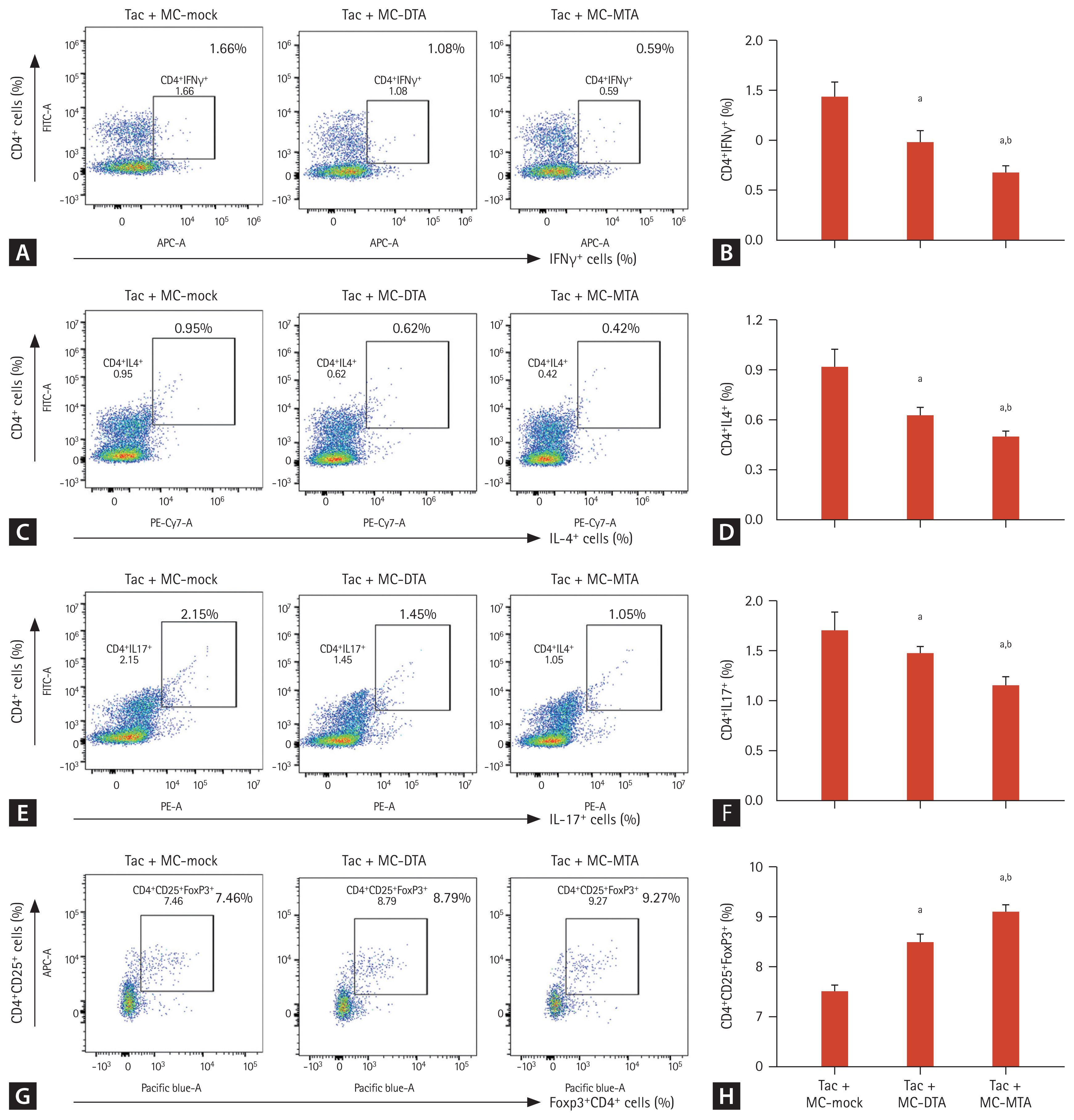
Consistent with the graft survival time results, fewer infiltrating cells were detected in the animals injected with Tac + MC-MTA than in those treated with Tac + MC-DTA (Fig. 9D). Flow cytometry also showed that the numbers of CD4+IFN╬│+, CD4+IL4+, and CD4+IL17+ cells were significantly lower in the Tac + MC-MTA group than in the Tac + MC-DTA group (Fig. 10AŌĆō10F). The CD4+CD25+Foxp3+ cell population was significantly higher in the Tac + MC-MTA group than in the Tac + MC-DTA group (Fig. 10G and 10H).
DISCUSSION
We have previously suggested a platform system to promote the self-production of therapeutic protein drugs by the host itself [31]. In this study, we evaluated a new synthetic protein drug (anti-CD25 mAb, IL-10, and CXCR3) in the form of an antibody-cytokine fusion using this system. Injected multiple targeting MCs (MTA) secreted a bioactive form of anti-CD25 mAb, IL-10, and CXCR3 in vivo, which was detected in the liver tissue for approximately 40 days. Moreover, mice treated with MCs encoding MTA showed prolonged skin allograft survival time accompanied by improved histologic changes and immunologic regulation. These findings suggest that the MTA protein drug obtained by the MC platform is functionally active and relevant to allograft rejection.
In this study, we used the MC vector for a new protein drug development and verification. Developing protein agents at a small scale is nearly impossible owing to the high cost of their purification and functional characterization. MCs are smaller than conventional plasmids, increasing the probability of cell delivery. Moreover, MCs escape gene silencing and show sustained expression as a result of the unique methylation patterns in these vectors [12]. The present study revealed that in vivo MC expression was detected for 40 days and the levels of anti-CD25, IL-10, and CXCR3 showed a 5.9, 3.9, 2.6-fold increase, respectively, in the MC-MTA group compared with the conventional vector (PP-MTA) group. Taken together, the MC system is proposed as a novel platform for the early development of protein drugs.
We selected anti-CD25, IL-10, and CXCR3 as therapeutic targets in an allograft rejection model. Treatments using anti-CD25 antibody agents significantly lower acute rejection rates compared with induction therapy [19,20]. However, the immunosuppressive mechanism of anti-CD25 agents is the deletion of CD25-expressing cells, but it is also has the same effect on Treg cells (FOXP3+ and FOXP3-CD25+ T cells) [21ŌĆō23], because Treg cells are critical regulators of immune tolerance in transplantation [24,25]. IL-10 conjugation with anti-CD25 antibody compensated for the possibility of Treg downregulation by anti-CD25 during alloimmune response because anti-CD25 targets share the expression of CD25, including both conventional activated T cells and CD4+CD25+ T cells (Tregs) [41,42]. Induction of chemokines during allograft rejection is accompanied by the infiltration of leukocytes bearing the respective chemokine receptors [43]. Of these, CXCR3 is a dominant factor directing T cells into mouse skin allograft confirmed by highly up-regulated T cells in the spleen and allograft from recipientsŌĆÖ mouse after transplantation [26]. Based on these findings, we designed MTA structure by the fusion of the anti-CD25 antibody and the dimer form of IL-10 via short a flexible linker, (G4S1)3 linker. CXCR3 was connected with IL-10 using (G4S1)2 linker as shown in Fig. 1.
Next, we examined whether the MTA produced by MCs was produced at sufficient levels to alleviate allograft rejection in vivo. We used a highly immunogenic murine skin allograft model and found that the MTA prolonged graft survival compared with MC-mock treatment, accompanied by improved histologic changes and immunologic regulation. We further investigated whether the MTA is effective due to CXCR3, which plays an important role in the migration towards injured or inflamed areas, compared with DTA encoding anti-CD25 and IL-10. First, we performed the in vitro transwell migration assay using ligands of CXCL (e.g., CXCL9, 10, 11) and revealed increased migrating cells under CXCL treatment. Second, we tested whether protein drugs were indeed detected around the graft area in the MC-MTA group using MC-MTA vector containing luminescence or RFP fluorescence. Furthermore, the MC-MTA group showed prolonged skin allograft survival, suppression of proinflammatory T cell subset, and induction of regulatory T cells compared with the MC-DTA group, suggesting that CXCR3 may be associated with skin allograft survival.
In clinical practice, combination therapy with a calcineurin inhibitor (CNI) and basiliximab is effective for preventing acute rejection early after KT [20], and is associated with a lower rate of adverse events than the lymphocyte-depleting induction [44]. We found a prolonged skin allograft survival time accompanied by improved histological changes and immunological regulation in the MC-MTA + Tac group compared with the MC-DTA + Tac group. CNI also blocks the T cell receptor-mediated production of IL-2 [45,46] and thus acts synergistically with IL-2 receptor antibodies to limit IL-2-mediated T cell proliferation [47,48]. Moreover, CD25 is upregulated by IL-2 [49,50], and CNI therapy inhibits the activation of marker expression on T cells, including CD25 [50]. Furthermore, enhanced migration function of MTA by CXCR3 may contribute towards promoting the above immunologic benefits. It is therefore conceivable that more intensive immunosuppression and immune tolerance may contribute towards high allograft survival by the combination of MC-MTA and Tac.
The delivery method using the MC system in this study is similar to conventional gene therapy using viral vectors (i.e., plasmid). Previous studies have investigated the delivery of MCs encoding natural inhibitor molecules [13,15,16]. In this reports, efficient tissue-target gene delivery is important for disease treatment by gene addition correction or knock-down. However, our approach is conceptually different from these approaches. MCs do not require targeting of the vector to the injured tissue or cells (as do current gene therapeutic strategies) because the circulating protein products (MTA) produced by host cells (i.e., liver cells) that take up the MCs treat the disease rather than the manipulation of the host genome in injured cells.
Although the current protocol was effective for the treatment of a mouse skin allograft rejection model, multiple improvements are needed for clinical applications. Clinically acceptable DNA delivery method is required to replace the hydrodynamic injection technique. The jetPEI (Polyplus-transfection SA., Illkirch, France) transfection reagent has already been tested in clinical trials [51], but the drugsŌĆÖ therapeutic levels cannot be obtained while simultaneously avoiding toxicity.
In conclusion, we developed a novel drug comprising anti-CD25/IL-10/CXCR3 using the MC technology system and confirmed its feasibility as a therapeutic tool for preventing allograft rejection in a mouse model. The MC system for synthetic protein drugs is now an emerging concept and may be promising for its cost effectiveness and convenience. We hope that the MC vector system may be useful for the development of novel biological drugs avoiding manufacturing or other practical problems.
KEY MESSAGE
1. We developed a novel drug comprising anti-CD25/ interleukin-10 (IL-10)/C-X-C motif chemokine receptor 3 (CXCR3) in the form of an antibody-cytokines fusion using the minicircle technology system.
2. Self-produced anti-CD25/IL-10/CXCR3 by the host were functionally active and reduced allograft rejection in a mouse model.
3. The minicircle vector system may be useful for developing new biological drugs, avoiding manufacturing or practical problems.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement 1
Supplement 1 Print
Print





