


INTRODUCTION
Liver fibrosis results from chronic injuries and often progresses to cirrhosis, liver failure, and hepatocellular carcinoma, and recent studies have suggested that correcting the etiologies improves hepatic fibrosis or cirrhosis [1–3]. However, complete reversal of liver fibrosis or cirrhosis is rare. Therefore, elucidating the pathways of fibrosis to develop anti-fibrotic therapy is important to halt the progression of hepatic fibrosis and reverse liver cirrhosis.
Liver fibrosis is characterized by the trans-differentiation of primary hepatic stellate cells (HSCs) [4]. Damage to hepatocytes causes an inflammatory reaction, which activates HSCs to become myofibroblasts that accumulate in the injured liver. One of the morphological characteristics of liver fibrosis is the deposition of extracellular matrix proteins. Several recent studies have suggested that the apoptosis of activated HSCs is responsible for mediating cell loss during recovery from fibrosis [5,6]. Hence, HSCs could be a major target for anti-fibrotic therapy. New therapies, including treatment using the anti-transforming growth factor β1 (TGF-β1) receptor monoclonal antibody fresolimumab, the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase inhibitor GKT137831, and an inhibitor of lysyloxidase-like-2, for treating liver fibrosis currently target the steps in HSC activation [7–9]. However, although completed trials have revealed the potential of emerging drugs for ameliorating hepatic fibrosis, appropriate strategies are still needed to limit it to the fibrotic environment. Moreover, the development process for a new clinical compound is complex and time taking. The duration of drug development may be shortened by repurposing pre-existing drugs for different clinical indications. Several drugs, such as statins and cyclooxygenase-2 (COX-2) inhibitors, have been reported to have anti-fibrotic effects. Statins inhibit the activity of hydroxymethylglutaryl-coenzyme A reductase, a key enzyme in cholesterol synthesis. Several clinical studies have shown that statins might offer clinical benefits in the setting of liver disease, including nonalcoholic steatohepatitis, cholestatic liver disease, hepatocellular carcinoma, and portal hypertension [10–13].
COX is an important enzyme involved in prostaglandin production. COX-2 plays a role in inflammation and carcinogenesis, and the COX-2/prostanoid pathway is associated with various liver diseases [14]. Activated HSCs induce COX-2 expression, suggesting that the COX-2/prostanoid pathway is involved in hepatic fibrosis as well [15]. In addition, several studies have shown that COX inhibitors have a pro-apoptotic effect on HSCs.
The combination of a statin and COX-2 inhibitor has the potential to exhibit a synergistic effect and decrease adverse effects. Therefore, further experimental investigations are needed to determine whether statin (simvastatin) and COX-2 inhibitor (NS-398) combined therapy exerts a synergistic effect on hepatic fibrosis or is toxic to cells.
METHODS
Culture and treatment of cells
The immortalized human hepatic stellate cell line LX-2 was donated by Dr. Scott L. Friedman (Mount Sinai School of Medicine, New York, NY, USA). LX-2 cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin, and streptomycin at 37°C in a humidified air incubator containing 5% CO2. Hepa RG cells are terminally differentiated hepatic cells derived from a human hepatic progenitor cell line that retains many characteristics of primary human hepatocytes. Hepa RG cells were purchased from Invitrogen (Carlsbad, CA, USA). The cells were maintained in William’s E medium supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 5 μg/mL insulin, and 5 × 10−5 M hydrocortisone hemisuccinate. Simvastatin and NS-398 were dissolved in distilled water at various concentrations and added to the cell culture medium for 24 and 48 hours.
WST-mediated cell viability assay in LX-2 and Hepa RG cells
The LX-2 and Hepa RG cells were seeded in 96-well plates at an initial density of 1 × 104 cells/well. The cells were rendered quiescent at 40% to 60% confluence by incubating them for 24 hours in a growth medium deficient in serum. The effect of simvastatin and NS-398 on the proliferation of the two cell types was evaluated, and cells cultured in serum-free medium were used as a control. Cell proliferation was determined using a WST assay (Wako, Osaka, Japan).
Measurement of DNA synthesis by bromodeoxyuridine incorporation enzyme-linked immunosorbent assay
Cellular DNA synthesis was determined by estimating the amount of bromodeoxyuridine (BrdU) incorporated into DNA using a colorimetric immunoassay (Roche Diagnostics GmbH, Mannheim, Germany). The protocol is described in the Supplementary Materials.
Annexin V/propidium iodide apoptosis assay
Annexin V/propidium iodide (PI) staining was conducted to determine the percentage of apoptotic cells in the total cell population. After simvastatin and NS-398 treatment, cells were collected and resuspended in a binding buffer (BD Pharmingen, BD Biosciences, San Jose, CA, USA). The cells were then incubated with 10 μg/mL PI and Annexin V (BD Biosciences), and the fluorescence intensity was determined using a FACSCalibur flow cytometer and BD CellQuest Pro software version 5.2.1 (BD Biosciences).
Animals and experimental protocol
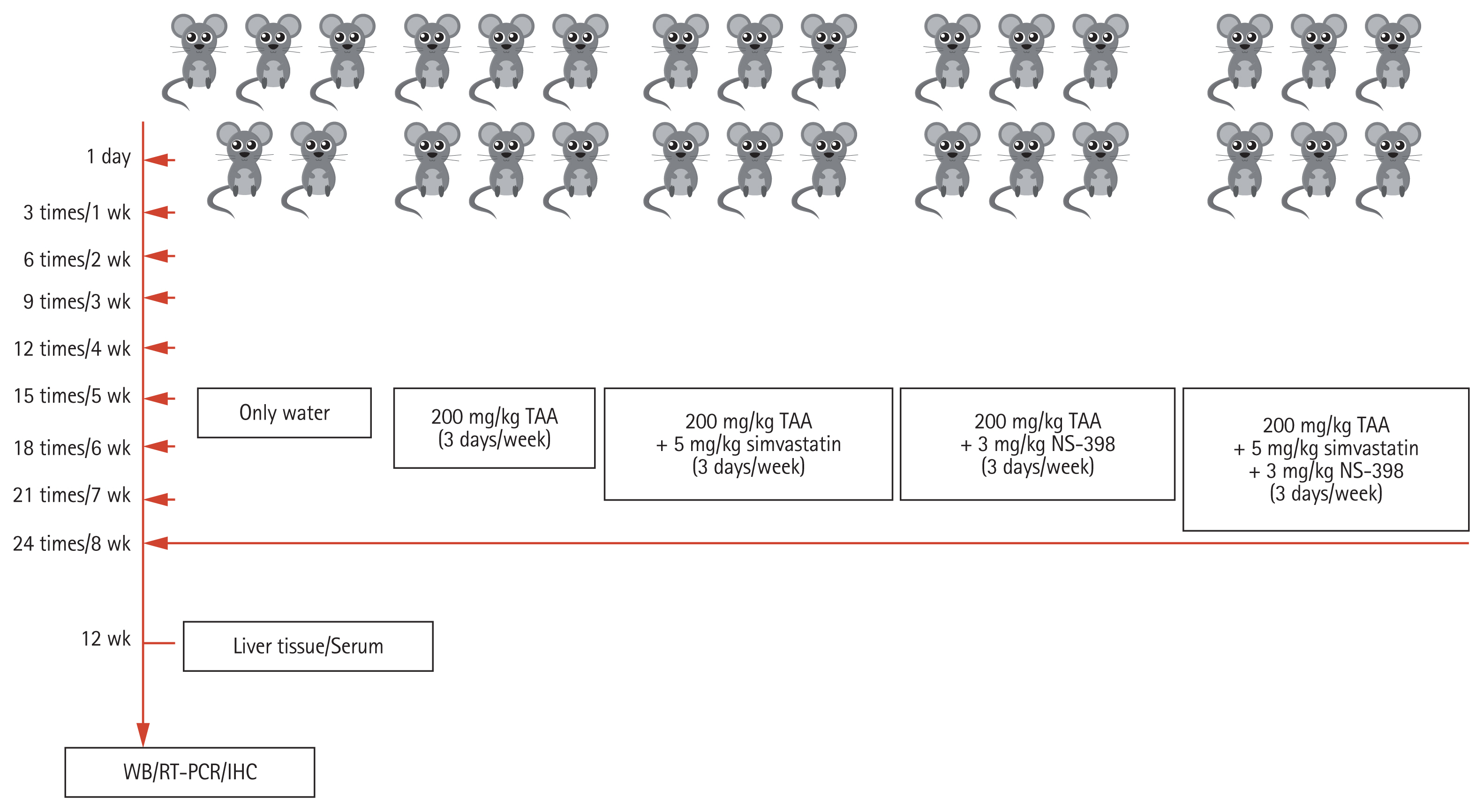
All animal experiments were performed with the permission of the Animal Care and Experimentation Committee of the Korea University Ansan Hospital (KULACUC-2015-69). Institute of Cancer Research (ICR) mice (body weight 15 to 20 g) were purchased from Orient Bio (Seongnam, Korea). The mice were maintained in a climate-controlled (21°C) room with less than 12-hour light-dark cycles and were given tap water and standard laboratory chow. Thioacetamide (TAA) was purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in sterile saline. Hepatic fibrosis was induced by administering 200 mg/L TAA to the drinking water of the ICR mice for 8 weeks. The ICR mice were randomly divided into five groups: (1) water (negative control); (2) 200 mg/kg TAA + saline (3 days/week); (3) 200 mg/kg TAA + 5 mg/kg simvastatin (3 days/week); (4) 200 mg/kg TAA + 3 mg/kg NS-398 (3 days/week); and (5) 200 mg/kg TAA + the combination of 5 mg/kg simvastatin and 3 mg/kg NS-398 (3 days/week) and sacrificed after 4 weeks (Fig. 1). The negative control group was not treated with TAA, simvastatin, or NS-398 but received an equal volume of normal saline. At the end of the experiment, serum was prepared and stored at −70°C until analysis of biochemical parameters was conducted. Liver tissues were used to determine hydroxyproline content, examine histology, and analyze mRNA and protein levels.
Western blotting
Protein expression was measured using a Western blot analysis. Proteins were extracted, and approximately 40 μg/mL of total protein was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and incubated with primary antibodies against α-smooth muscle actin (α-SMA), collagen type I, pro-caspase 3, extracellular signal-regulated kinase (ERK) 1/2, phospho-ERK1/2, Bax, Bcl-2, TGF-β1, Smad2, phospho-Smad2 (Ser465/467), Smad3, phospho-Smad3 (Ser423/425), tissue inhibitor of matrix metalloproteinases (TIMP)-1, TIMP-2, matrix metalloproteinase (MMP)-1, and MMP-13 (Supplementary Table 1). β-Actin was used as the loading control.
Quantification of collagen content by hydroxyproline assay
Total collagen content was determined by quantifying hydroxyproline content according to the manufacturer’s instructions (BioVision Inc., Milpitas, CA, USA). A brief protocol is provided in the Supplementary Materials.
RNA extraction and real-time polymerase chain reaction
Total RNA was isolated from frozen liver tissue using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions (see Supplementary Information). Specific primers for TIMP-1, TIMP-2, MMP-1, and MMP-2 were designed from their GenBank sequences and synthesized by Bio Basic Inc. (Markham, ON, Canada) (Supplementary Table 2).
RESULTS
Safety of using simvastatin and NS-398 in liver cells
Influence of simvastatin and NS-398 on the viability of HSCs and Hepa RG cells
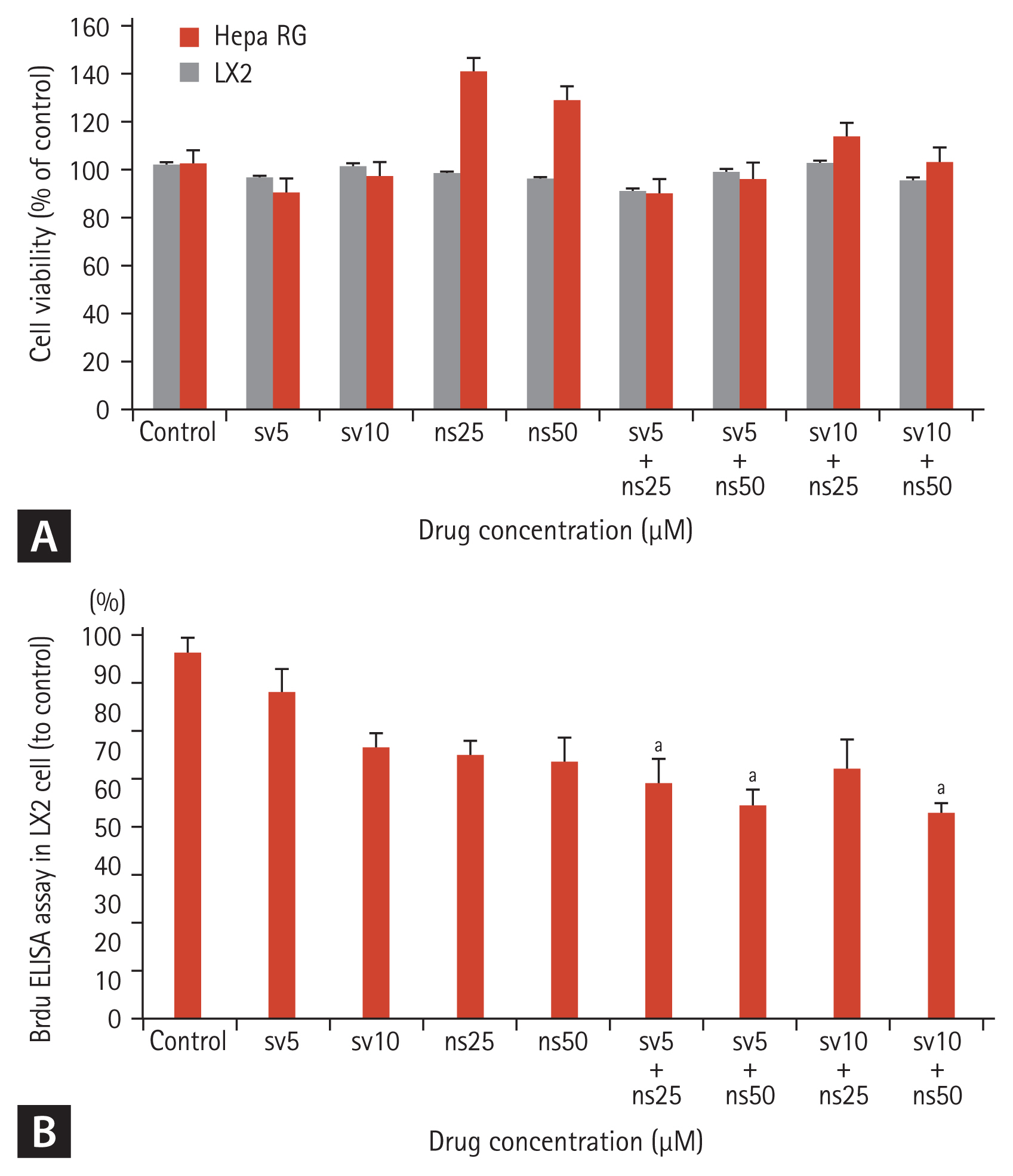
We evaluated the effects of various concentrations of simvastatin and NS-398 on the viability of LX-2 and Hepa RG™ cells. The cells were incubated with simvastatin (0, 5, or 10 μM) with or without NS-398 (0, 25, or 50 μM) for 24 hours. A cell proliferation reagent (WST-1) was used to measure cell toxicity according to the manufacturer’s protocol. Simvastatin (< 10 μM) or NS-398 (< 50 μM) exhibited no cytotoxic effects. Additionally, no definite cytotoxic effect was detected with the combination of simvastatin and NS-398 (Fig. 2A).
Combined treatment with simvastatin and NS-398 induced improved anti-fibrotic effects on an HSC cell line and a mouse model
Effect of simvastatin and NS-398 on apoptosis
Apoptosis was induced to determine the possible mechanism of action of simvastatin and NS-398 at various concentrations in LX-2 cells. These observations were confirmed by Annexin V/PI staining of cells treated with the indicated drugs (Supplementary Fig. 1). The ratio of apoptotic cells at 5 and 10 μM simvastatin and 25 and 50 μM NS-398 were 1.15%, 1.49%, 3.52%, and 4.48%, respectively. This value increased to 8.44% after combined treatment with 5 μM simvastatin and 50 μM NS-398 a nd was highest at 27.3% after combination treatment with 10 μM simvastatin and 50 μM simvastatin. These findings demonstrate that combination therapy using simvastatin and NS-398 enhanced apoptosis in a dose-dependent manner compared to monotherapy.
Simvastatin and NS-398 combination further inhibited proliferation of activated HSCs
LX-2 cells were treated with different concentrations of simvastatin (0, 5, or 10 μM) with or without NS-398 (0, 25, or 50 μM) for 24 hours in a BrdU-proliferation assay. The proportions of proliferating cells at 5 and 10 μM simvastatin were 89.6% and 74.8% of the control value, respectively (Fig. 2B). NS-398 also inhibited cell proliferation. The proportions of proliferating cells treated with 5, 10, and 25 μM NS-398 were 98.5%, 77.0%, and 72.6% of the control value, respectively. The combination of various concentrations of simvastatin and NS-398 resulted in a significant reduction (51%) in cellular proliferation compared to the control.
Increased inhibition of fibrogenesis by simvastatin and NS-398 combination
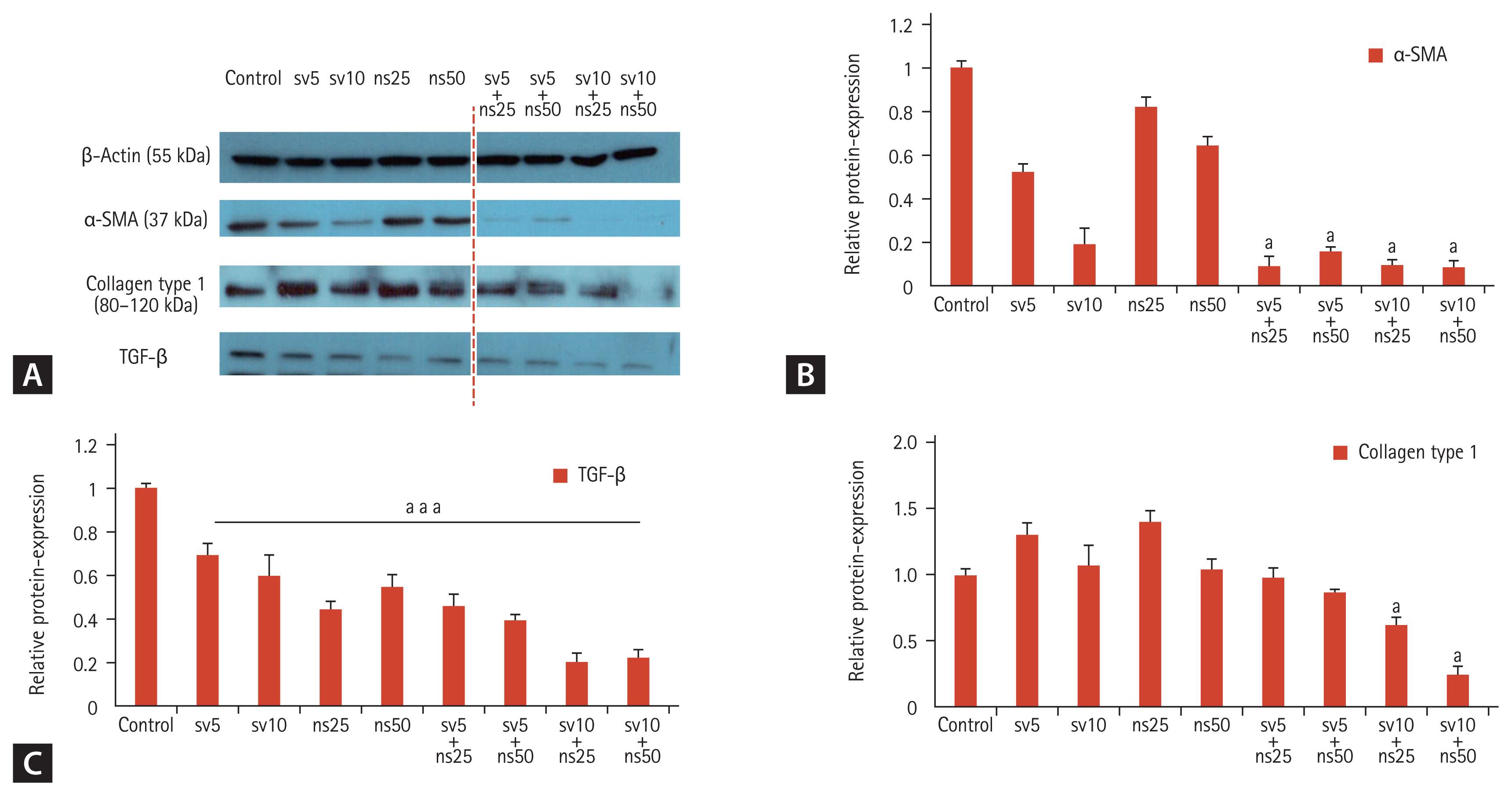
LX-2 cells constitutively express α-SMA and human collagen type-1. Simvastatin treatment reduced the expression of α-SMA, whereas NS-398 treatment had little effect on the expression of α-SMA and collagen protein (Fig. 3A and 3B). However, the combination of simvastatin (10 μM) and NS-398 (25 or 50 μM) further inhibited fibrogenesis of HSCs as indicated by a reduction in the amounts of α-SMA and collagen type I proteins. In addition, the expression of TGF-β, which is a key mediator in the pathogenesis of liver fibrosis, was significantly lower in cells treated with the combination of simvastatin and NS-398 compared to the control (Fig. 3C). However, the levels of phosphorylated Smad2/3 proteins were not obviously affected despite their decreasing tendency with the combined simvastatin and NS-398 treatment (Supplementary Fig. 2).
Simvastatin and NS-398 combined treatment reversed hepatic fibrosis in a mouse model
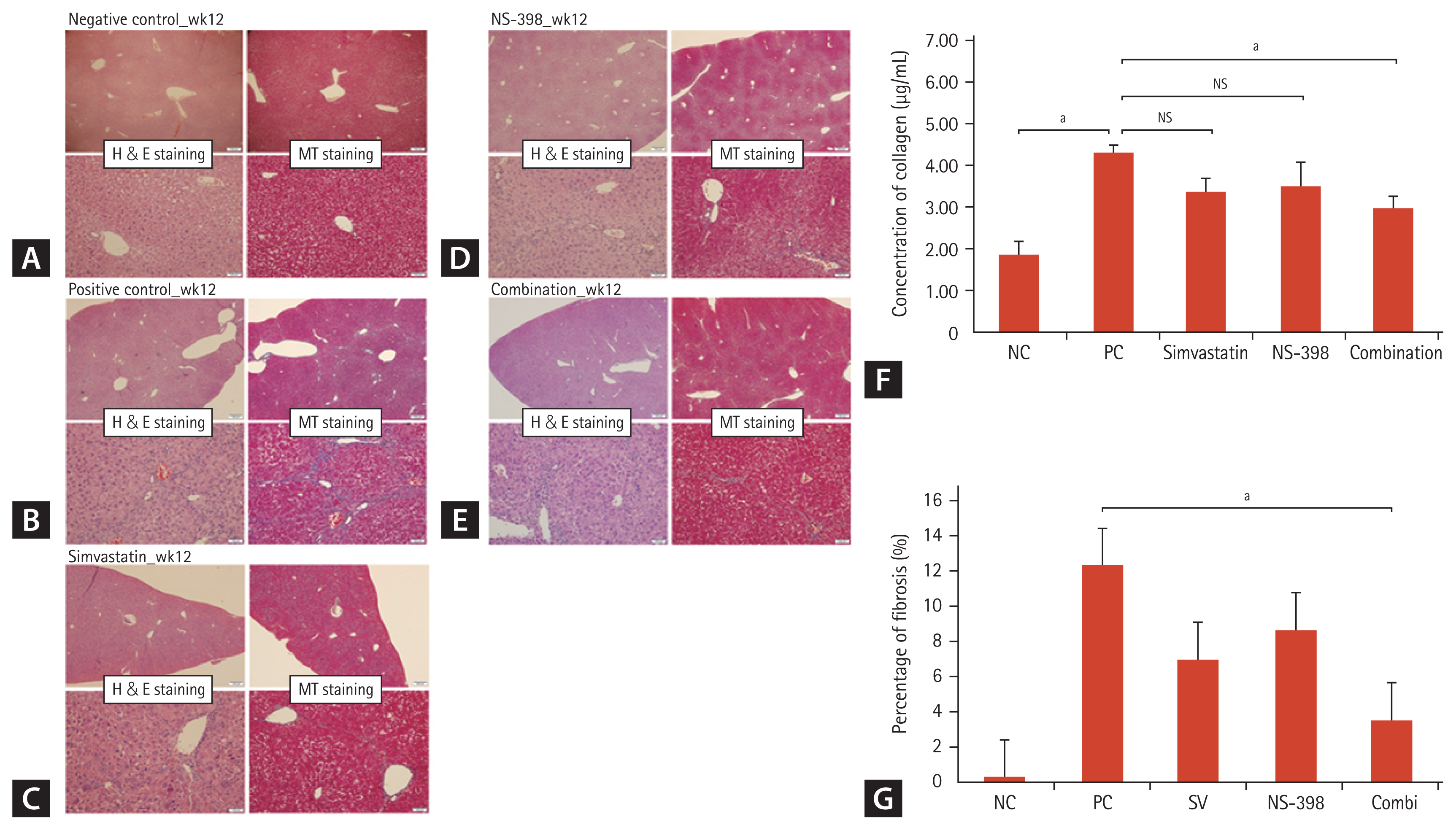
Administration of TAA for 8 weeks caused moderate to severe liver injury, as demonstrated by fibrosis with thick fibrotic septa and pseudo-lobular formation [16]. Treatment with simvastatin or NS-398 resulted in the attenuation of histological fibrosis (Fig. 4A–4E). The combined simvastatin and NS-398 treatment led to a significantly lower percentage of fibrosis than that with either agent alone. First, the percentage of fibrosis after Masson’s trichrome staining, as measured by morphometry, decreased to 3.51% after the combination treatment with simvastatin and NS-398 (Fig. 4F). Next, a significant decrease in histological fibrosis was detected in the combined treatment group using the hydroxyproline assay (Fig. 4G). The levels of aspartate aminotransferase and alanine aminotransferase increased in positive control mice; however, no difference was observed between the positive control and simvastatin or/and NS-398-treated mice (Supplementary Table 3).
Diverse cellular mechanisms are involved in the inhibition of fibrosis by the combination of simvastatin and NS-398
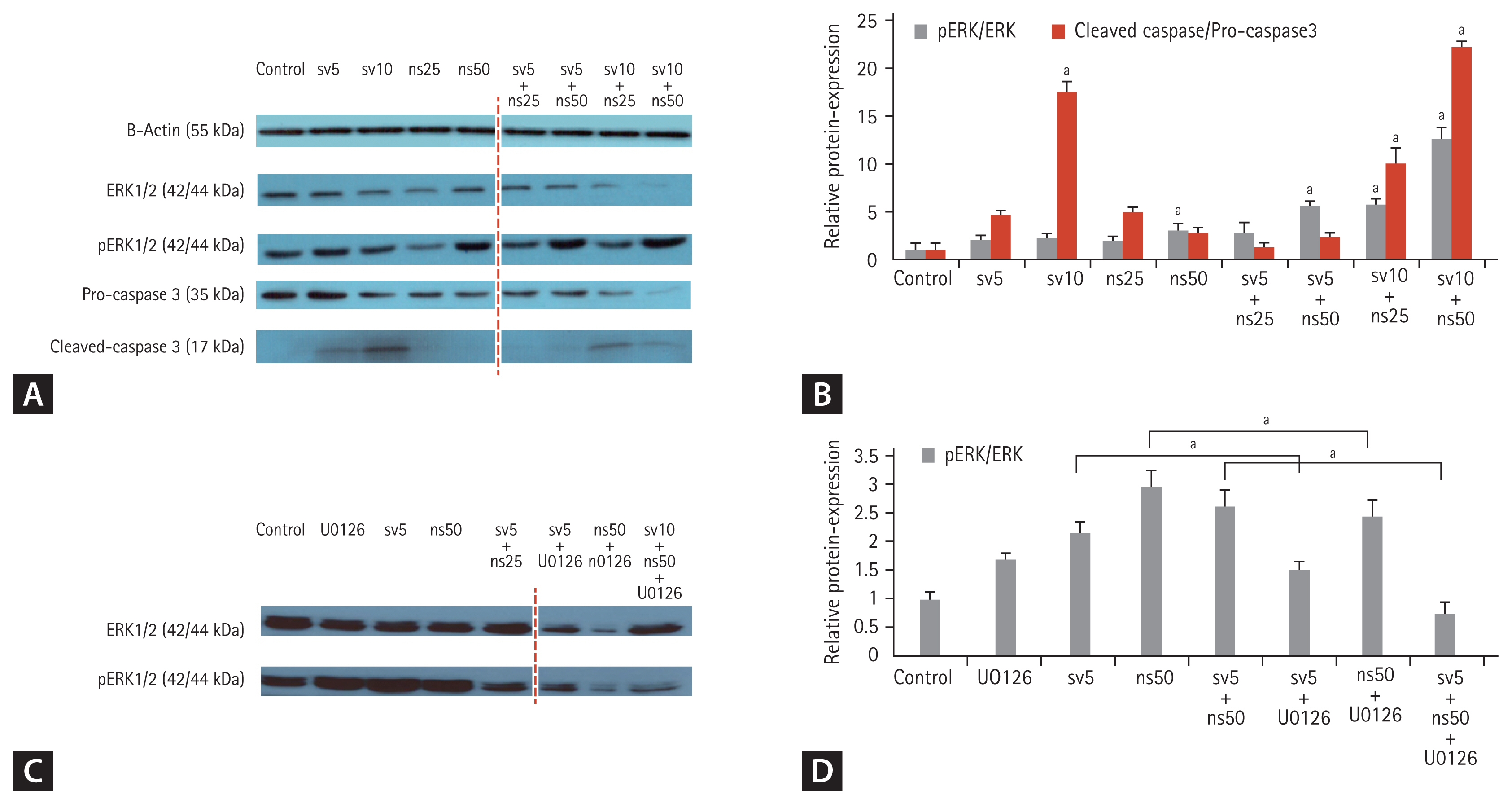
Simvastatin and NS-398 combined treatment- induced HSC apoptosis is primarily dependent on ERK activation
The effects of simvastatin and NS-398 on pro-apoptotic and anti-apoptotic pathways were evaluated in HSCs by the Western blot analysis, including that of ERK1/2 and phosphorylated-ERK1/2 (p-ERK1/2) (Fig. 5A and 5B). The results demonstrated that the expression levels of p-ERK1/2 were markedly increased with NS-398 treatment compared with that in cells incubated with simvastatin. Moreover, a significant increase in p-ERK1/2 expression levels was noted following the combined simvastatin and NS-398 treatment of LX-2 cells. Caspase-3 is activated by upstream proteases during apoptotic signaling events. Caspase-3 activity increased as a result of cleavage of procaspase-3 in the cells treated with the combination of simvastatin and NS-398 (Fig. 5A and 5B). The increase in caspase-3 activity was pronounced with 10 μM simvastatin. To further explore the role of ERK in the anti-fibrotic effects of simvastatin and NS-398, HSCs were treated with U0126, an ERK1/2 inhibitor, resulting in reduced phosphorylation of ERK1/2 (Fig. 5C and 5D). These data indicate that the combination of simvastatin and NS-398 induced apoptosis in HSCs, which may be related to the ERK-dependent pathway.
Bcl-2/Bax signaling pathway is affected by combined treatments using simvastatin and NS-398
We focused on Bcl-2/Bax signaling to study the potential mechanism by which the simvastatin and NS-398 combined treatment improved liver fibrosis. Bax initiates apoptosis by forming pores in the outer mitochondrial membrane [17]. The anti-apoptotic Bcl-2 protein inhibits the release of cytochrome c through mitochondrial pores. The Western blot analysis showed that the trends of the Bcl-2 and Bax expression levels in the total cell protein extracts were opposite to each other (Fig. 6). When compared to the levels in cells treated with NS-398 alone, the expression level of Bax was markedly increased after treatment with simvastatin, additionally. Treatment with the combination of simvastatin and NS-398 also upregulated Bax expression but downregulated Bcl-2 expression in HSCs. However, this effect was not blocked by the addition of the ERK1/2 inhibitor U0126.
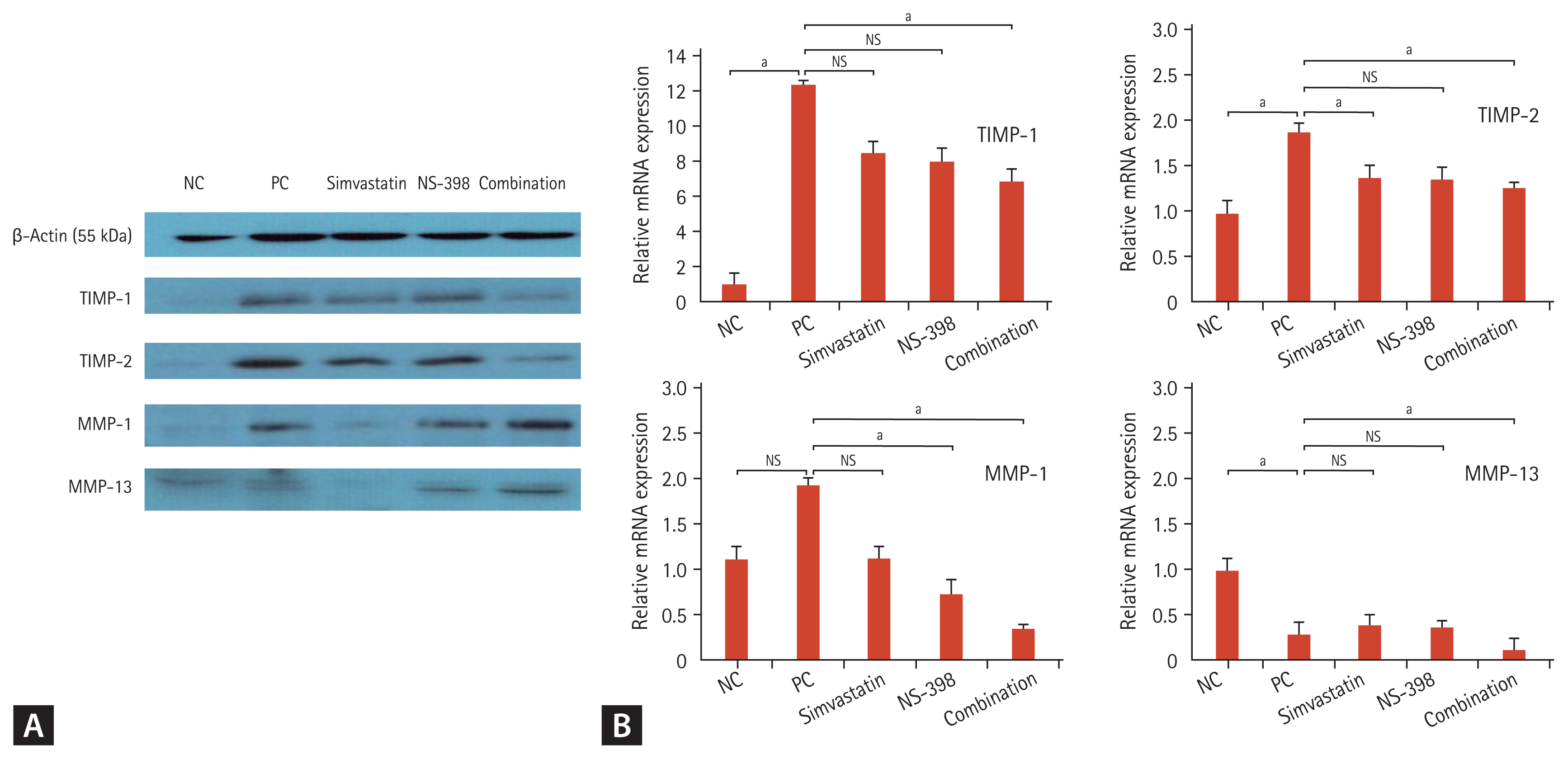
Anti-fibrotic effects of TIMP-1 and -2 on HSCs are mediated by MMP-1 and 13
TGF-β regulates TIMP and MMP expression [18]. TIMP and MMP contribute to the progression and regression of liver fibrosis [19]. According to the Western blot analysis, the combination of simvastatin and NS-398 resulted in a greater decrease in TIMP-1 and TIMP-2 expression than either drug alone, indicating that apoptosis was activated more easily with the combined treatment than with either drug alone in mice liver tissue (Fig. 7A). The same effect was observed in mRNA analysis (Fig. 7B). TIMP-1 and TIMP-2 levels significantly decreased in mice treated with the combined simvastatin and NS-398 treatment according to real-time polymerase chain reaction findings.
The combination of simvastatin and NS-398 resulted in increased MMP-1 and MMP-13 expression in mice with TAA-induced fibrosis according to the Western blot analysis. However, MMP-1 and MMP-13 mRNA levels decreased in the combined group, contrary to the findings of the Western blot results. MMP-13 mRNA expression was lower in positive control mice with TAA-induced fibrosis, and MMP-13 mRNA expression was further decreased in the combined simvastatin and NS-398 group.
DISCUSSION
In this study, the combination of simvastatin and NS-398 had a synergistic anti-fibrotic effect on LX-2 cells and TAA-treated mice. Collagen-related proteins decreased significantly in HSCs treated with the combination of simvastatin and NS-398. In addition, histological improvement in fibrosis was observed in the mice.
ERK1/2 signaling cascades strongly stimulate collagen production by HSCs [20]. The ERK pathway plays a major role in cell proliferation and differentiation. Hence, it appears to play a crucial role in the mitogenic effect of HSCs. A previous study reported that statins induce apoptosis in activated HSCs by increasing the phosphorylation of ERK1/2 [21]. One study reported that all COX inhibitors increase the phosphorylation of ERK1/2 [22]. In our study, the combination of simvastatin and NS-398 markedly increased the phosphorylation of ERK1/2. However, there was no difference between the control and 25 μM NS-398, 5 μM simvastatin, 5 μM simvastatin + 25 μM NS-398, 10 μM simvastatin, and 10 μM simvastatin + 25 μM NS-398, respectively. These results are thought to be caused by NS-398 having a greater influence on the ERK pathway than simvastatin. In particular, the increase of p-ERK level was more remarkable with 50 μM NS-398 compared to that with 25 μM NS-398. A previous study reported that celecoxib (a COX-2 inhibitor) induced apoptotic cell death at a concentration > 50 μM in human HSCs; however, NS-398 at even higher concentrations (100 μM) had no effect on cell viability. In this study, NS-398 monotherapy had no significant effect on cell proliferation or fibrosis. However, when > 50 μM of NS-398 and simvastatin were combined, apoptosis of activated HSCs was upregulated through the ERK pathway. Cellular ERK activation inhibits or enhances apoptosis in some cells [23]. Therefore, the role of ERK activation may differ depending on the cellular results. Diverse apoptotic signals come together at the mitochondrial level [24]. Cytochrome c is released from mitochondria following a loss of mitochondrial transmembrane potential. In our study, the release of cytochrome c was induced by ERK activation, followed by an increase in caspase-3 pathway activity. Hence, we have shown that the combination of simvastatin and NS-398 leads to apoptosis in activated HSCs by ERK activation, initiating the caspase cascade.
Statins exhibit anti-fibrotic properties against fibrotic diseases of the kidney, heart, lung, and skin by modulating TGF-β signaling [25–27]. In addition, statins inhibit the angiotensin II/Smad pathway. TGF-β/Smad signaling is a key pathway that leads to liver fibrosis [28,29]. However, a recent study reported that prostaglandin E2 (PGE2) inhibits TGF-β-induced collagen synthesis and NS-398 suppresses PGE2 synthesis, resulting in increased levels of collagen-α in HSCs. In our study, NS-398 alone had little effect on the expression of collagen proteins. However, the combined simvastatin and NS-398 treatment reduced the levels of α-SMA and collagen type I proteins. In addition, TGF-β was inhibited by the combination of simvastatin and NS-398. Although it is unclear whether statin or NS-398 caused the main effect, it is suggested that the combined simvastatin and NS-398 treatment inhibited TGF-β signaling, resulting in counter-regulation of the profibrotic effect on HSCs.
Considering that Bcl-2/Bax signaling plays a key role in the mitochondrial apoptosis pathway and is an important regulator of intrinsic apoptosis, we studied the anti-apoptotic effects of simvastatin and NS-398 by investigating Bcl-2 and Bax protein levels. Importantly, apoptosis was induced by downregulating Bcl-2 expression and upregulating Bax and caspase-3 expression through the Bcl-2/Bax signaling pathway. Loss of mitochondrial membrane potential due to changes in mitochondrial membrane permeability causes an increase in the Bax/Bcl-2 ratio, which is followed by the release of cytochrome c and activation of caspases-9 and −3. The apoptosis initiator caspase-8 induces cleavage of Bid, increasing the Bax/Bcl-2 ratio and directly activates caspase-3. The present data are consistent with this hypothesis. The combination of simvastatin and NS-398 was shown to induce apoptosis in HSCs by activating the mitochondrial pathway and the Bcl-2/Bax pathway.
Drugs that induce apoptosis in HSCs can cause liver fibrosis to regress [30]. Increased collagenase activity is the primary pathway involved in the resolution of fibrosis. Increased activity of collagen-degrading enzymes (e.g., MMPs) is associated with decreased TIMPs. A previous study demonstrated that TIMP-1 directly inhibited HSC apoptosis in vitro [31]. Moreover, statins significantly reduced TIMP-1 and TIMP-2 mRNA levels and attenuated liver fibrosis in choline-deficient L-amino acid-defined rats [32]. In our study, compared to monotherapy with simvastatin or NS-398, the combination these drugs resulted in a greater decrease in TIMP-1 and TIMP-2 levels in both Western blot and mRNA analyses. Inhibition of TIMP-2 was more prominent than that of TIMP-1 in our study. This result corresponds to a previous in vitro study that showed that LX-2 cells expressed only a small amount of TIMP-1 protein [33]. TIMP-1 and −2 may be important regulators of apoptosis. However, several studies have reported that the anti-apoptotic effect of TIMP-1 is independent of its ability to inhibit MMP activity [31]. There was a discrepancy in the interaction between TIMP and MMP in our study. An increase in MMP-1 and MMP-13 activity was observed in mouse tissue with improved fibrosis. However, the MMP-1 and MMP-13 mRNA results were different from those of protein. One study demonstrated that TIMP-1 inhibits apoptosis of activated HSCs by inhibiting MMP-1, resulting in fibrosis in vitro [31]. Another study reported that overexpression of TGF-α may attenuate hepatic fibrosis, in part, because of upregulated MMP-1 expression [34]. Similarly, the role of MMP-13 in the anti-fibrotic pathway remains controversial [35]. The reason for this discrepancy is not clear, but these contradictory results can be explained by examining the dynamic interaction between MMP and TIMP. Previous data have shown that there may be no correlation between MMP mRNA expression and enzymatic activity probably because of blocked activation of the MMP pathway or the formation of complexes with TIMP [36]. Moreover, statins may impact DNA methylation, which is a key regulator of gene expression. MMP enzyme activity itself may be affected by methylation inhibition, which may cause a discrepancy between the protein and RNA results.
Although the combination therapy was attempted as an anti-fibrotic treatment in liver fibrosis and showed improvement in histological improvement in vivo through simvastatin and NS-398 combination treatment, the mechanism that can explain these complex observations was poorly investigated, especially that of NS-398 treatment. It would have been possible to provide more details on the mechanisms if we analyzed the upstream regulator/signaling cascade of ERK and other compensatory pathways (such as autophagy pathway or phosphoinositide 3-kinase/Akt). A complex interplay exists between hepatocytes and HSCs during hepatic fibrogenesis. Recently, many studies have reported that a co-culture model may be useful for mimicking the liver’s microenvironment. Regarding the expression of fibrotic markers, we only analyzed them in HSCs, but in order to analyze the association between cells, more consistent results may have been derived if we experimented with a simultaneous co-culture model of hepatocytes and HSCs.
In the present study, the combination of simvastatin and NS-398 produced significant inhibition of cell proliferation and attenuation of liver fibrosis, while treatment with either simvastatin or NS-398 alone only had a moderate effect on HSCs and liver tissue. In addition, no toxic effects of the combined simvastatin and NS-398 treatment were observed. The combination of simvastatin and NS-398 had the greatest effect on apoptosis in the present study, and this effect was associated with multiple pathways, including the ERK pathway, followed by an increase in the caspase cascade, inhibition of TGF-β signaling, an increased Bax/Bcl-2 ratio, and inhibition of TIMP-1 and TIMP-2. Clinically, the combination of these two drugs could be suggested as a promising therapeutic strategy to maximize pharmaceutical efficacy and minimize adverse events in patients with liver fibrosis. Further detailed studies are warranted to investigate the molecular mechanisms involved in the synergistic effects of simvastatin and NS-398 and their clinical efficacy.
KEY MESSAGE
1. A combination treatment using simvastatin and a cyclooxygenase-2 inhibitor, NS-398, produced greater inhibition of hepatic stellate cell proliferation and attenuation of liver fibrosis compared with treatment with either drug alone.
2. These anti-fibrotic effects were associated with multiple pathways, including the extracellular signal-regulated kinase pathway, Bax/Bcl-2 signaling, and tissue inhibitor of matrix metalloproteinases 1 (TIMP-1) and TIMP-2 protein expression.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement 1
Supplement 1 Print
Print





