


CONSENSUS SUMMARY
INTRODUCTION
Familial hypercholesterolemia (FH; indicating a heterozygous form in this article unless otherwise defined) is the most common monogenic hereditary disorder with an autosomal dominant pattern. The reported frequency is approximately 1/200 to 1/500 [1,2]. However, regional variations may exist. The exact prevalence in Korea has not yet been reported, although it may be similar to that in other countries. FH cases have been reported since the 1980s in Korea [3,4].
LDLR, APOB, and PCSK9 are common targets of pathogenic variants (PVs), and there are various mutations in each gene. Other rare PVs have been reported. A substantial number of patients with a phenotype compatible with FH show no PVs. Therefore, even though it is a hereditary disease, clinical diagnostic criteria play an important role in its diagnosis. In general, there is no unified diagnostic standard, and various clinical criteria are used in each country. Cardiovascular risk is elevated up to 10 times and complications such as coronary artery disease (CAD) occur earlier than in the general population [5]. Early aggressive lipid-lowering can reduce cardiovascular risk in patients with FH. Therefore, active screening for early diagnosis is critical. Considering the distribution of low-density lipoprotein cholesterol (LDL-C) levels in Koreans, a large number of individuals are expected to be diagnosed with FH based on the current cut-offs of major foreign criteria. However, there are limited data on Korean patients with FH, and awareness of FH among medical personnel and the public is very low. Therefore, the aim of this article was to provide current epidemiological data of FH in Korea and increase awareness, interest, and early detection rate of this disease. In addition, the authors summarized expert consensus to guide appropriate and updated treatment of FH. The authors have obtained informed consent from patients.
CLINICAL AND GENETIC CHARACTERISTICS OF FH
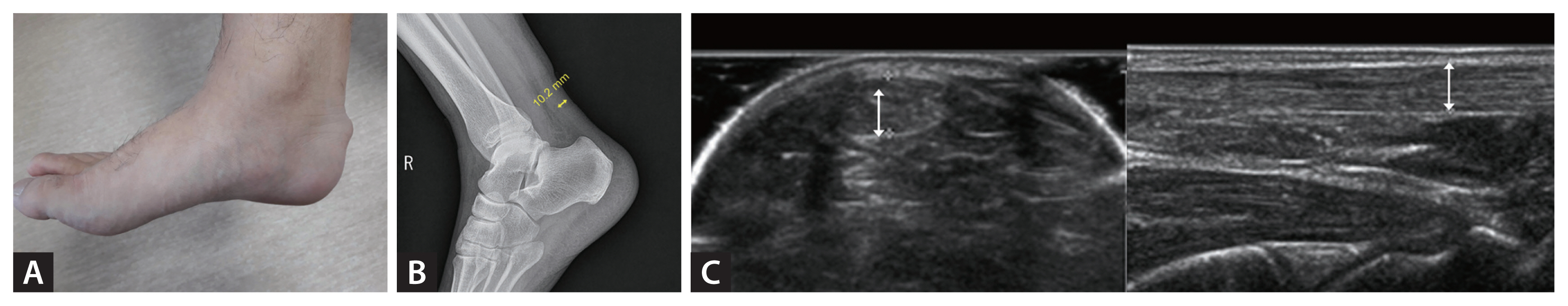
Typical physical findings are tendon xanthoma (Fig. 1A) and premature arcus cornealis. However, their sensitivities are low, and many affected patients do not have these manifestations. In addition, thickened Achilles tendon can be revealed by lateral ankle X-ray (Fig. 1B). Briefly, the angle between the lower leg bone and sole should be 90 degrees. The imaging distance is 120 cm and the following imaging conditions need to be applied: 50 kV and 5.0 mAs [6]. Achilles tendon thickness and width can be measured by ultrasonography (Fig. 1C). The measurement is usually performed with ankles flexed at 90 degrees and images are obtained with horizontal and linear sections. The Korean FH registry 2020 demonstrated that clinically diagnosed patients with FH had median total cholesterol and LDL-C levels of 306 and 221 mg/dL, respectively, and the prevalence of tendon xanthoma and CAD was 20% and 19%, respectively. Among registered patients, 60%, 36%, and 3% had a family history of severe hypercholesterolemia, premature CAD, and tendon xanthoma, respectively [7].
CARDIOVASCULAR RISK OF FH
If untreated, patients with heterozygous FH (HeFH) show an elevated risk of CAD up to 10 times before the age of 55 (male) or 60 (female), particularly those with definite or probable types [5]. A study supported by the Korean Society of Lipid and Atherosclerosis analyzed a nationwide cohort of 2.3 million individuals. During a median follow-up of 6.1 years, those with LDL-C of 190–224, 225–259, and ≥ 260 mg/dL had up to 2.4-times higher risk of cardiovascular risk (myocardial infarction, coronary revascularization, or ischemic stroke) compared to those with LDL-C < 160 mg/dL. All-cause death occurred up to 2.3 times more frequently in individuals with LDL-C levels ≥ 190 mg/dL [8].
Estimating cardiovascular risk using well known risk calculators in patients with FH is inappropriate. LDL-C levels in these patients increase from a young age, and such calculations may lead to risk underestimation [9]. In this regard, the American guidelines for lipid-lowering therapy (LLT) classify individuals with LDL-C ≥ 190 mg/dL as a statin benefit group whose cardiovascular risk is eligible for pharmacological therapy [10]. The 2019 European guidelines classify patients with FH into very high -or high-risk groups according to the presence of other risk factors [5]. These include traditional factors, such as age, male sex, hypertension, smoking, LDL-C, history of atherosclerotic cardiovascular disease, and body mass index [11]. A Korean registry supported by the Korean Society of Lipid and Atherosclerosis similarly reported that hypertension and low high-density lipoprotein cholesterol (HDL-C) levels were predictors of clinical outcomes in patients with FH [12].
DIAGNOSIS OF FH
Clinical diagnosis
The Dutch Lipid Clinic Network Criteria are most commonly used globally (Table 1) [5], and the Simon Broome criteria are also frequently used (Table 2) [13]. Currently, diagnosing FH based on these criteria seems most reasonable. Both criteria include LDL-C levels, physical findings such as tendon xanthoma, family history of premature CAD or severe hypercholesterolemia, and DNA mutations. A few countries have their own criteria for clinical diagnoses. In Canada and Japan, they analyzed models including LDL-C levels, clinical findings, and family history, and determined criteria with optimal sensitivity and specificity [14,15]. The Korean FH registry 2020 revealed that the rates of PV carriers were 50% to 64% when the patients were classified as definite or probable types by the Dutch or Simon Broome criteria [7]. Not surprisingly, the sensitivity and specificity were inversely correlated according to the criteria used. Thus, it may be less effective to identify patients when definite-type diagnostic criteria are applied. It is preferable to use criteria with high sensitivity for patient screening, whereas it is appropriate to use criteria with high specificity for cascade screening [16]. It may be helpful to choose diagnostic criteria flexibly according to diagnostic purpose.
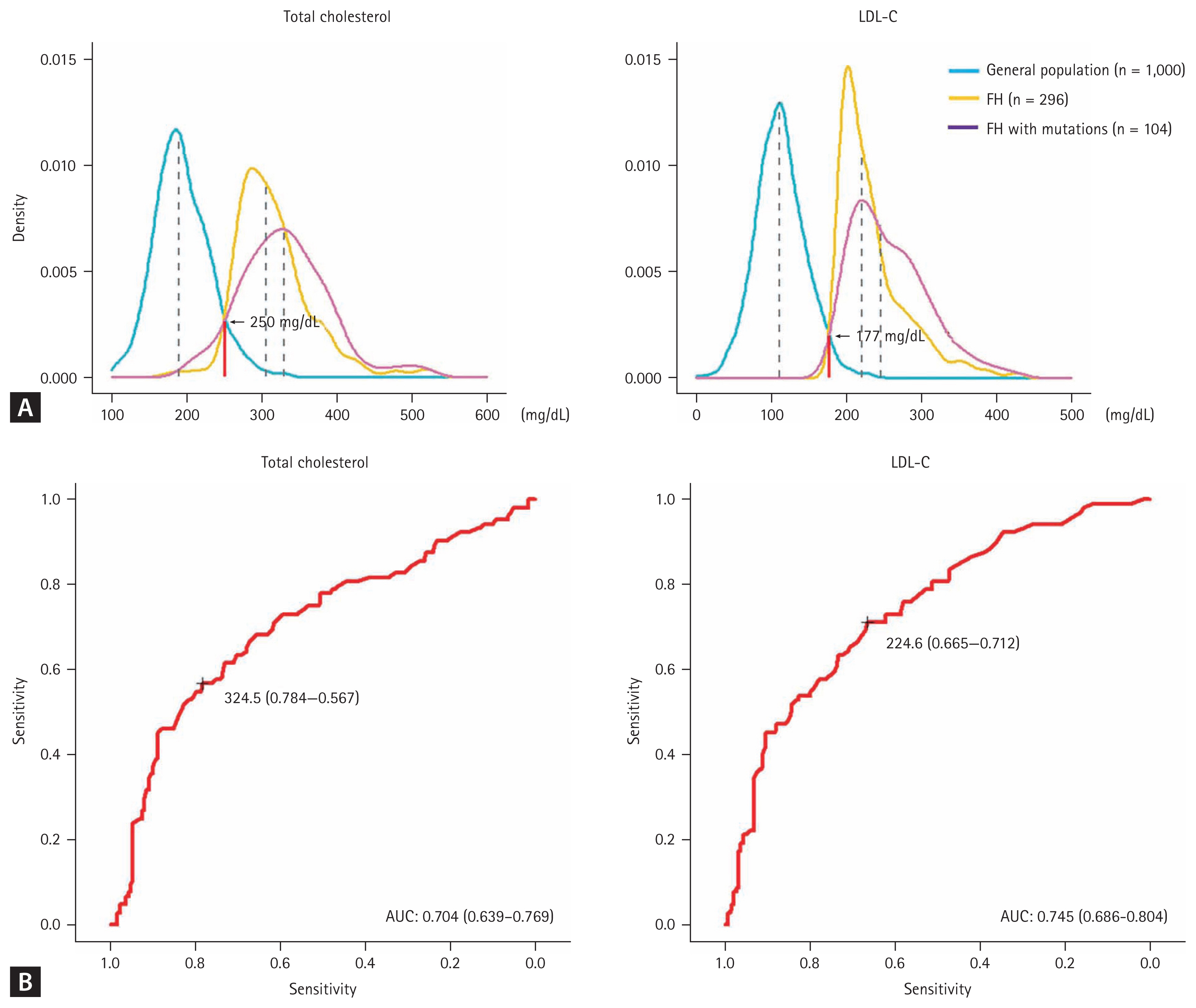
Currently, Korean data for the unique diagnostic criteria of FH are insufficient. However, the Korean FH registry 2020 analyzed LDL-C distributions in the general population and patients with FH and reported cut-off values of total cholesterol and LDL-C of 250 and 177 mg/dL, respectively (Fig. 3A). For PV carriers, the cut-off values of total cholesterol and LDL-C with optimal sensitivity and specificity were 325 and 225 mg/dL, respectively (Fig. 3B) [7]. Therefore, these values may be used when screening FH or cascade screening within a family in Korea.
Genetic diagnosis
Although FH is a genetic disease, genetic testing is performed on a small number of individuals with suspected FH. Genetic testing is beneficial for (1) clearer diagnosis, (2) refining risk assessment and subsequent promotion of LLT, and (3) making cascade screening more efficient [17]. A recent study demonstrated that the cardiovascular risk of PV carriers is three times higher than that of those with similar physical signs but without PVs [18]. As previously mentioned, the prevalence of PV carriers can vary according to the type of clinical diagnosis. FH diagnosis cannot be ruled out when a person has an FH-compatible phenotype and family history, even if PVs are not identified in genetic testing. Such cases may be of polygenic origin, technical problems, autosomal recessive PVs of the LDLRAP1 gene, or a variant of a novel gene.
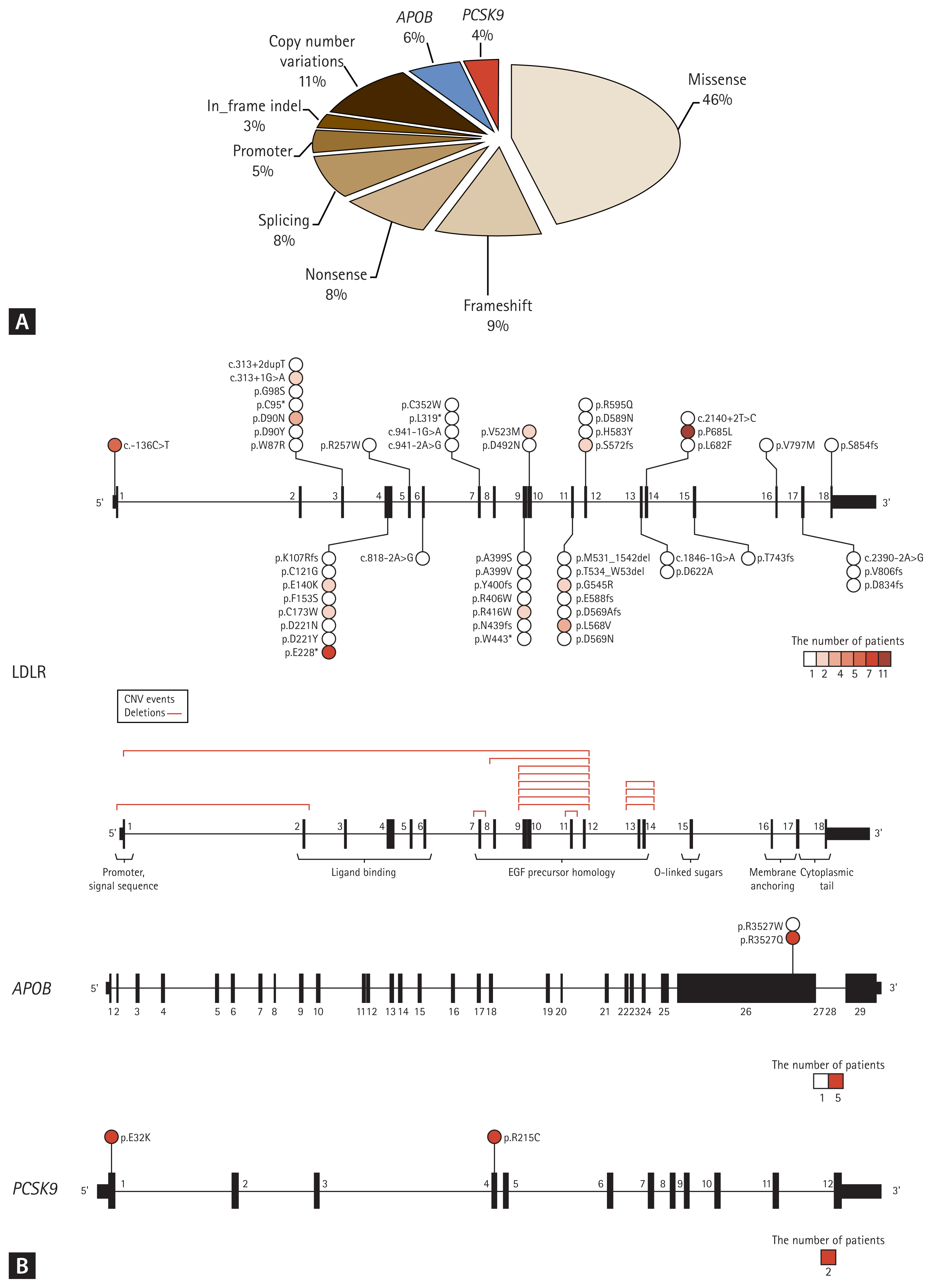
The target genes of FH genetic testing are LDLR, APOB, and PCSK9. Due to gradual cost reduction, many institutes recently examine these three genes using next-generation sequencing and deletion/duplication analysis. Similar to data from other countries, copy number variations are found in approximately 10% of PV carriers, but this type of PV requires multiplex ligation-dependent probe amplification or the TaqMan method for validation [7]. When a PV is identified, it is critical to properly interpret whether it is a causative variant. There are few databases on LDLR variants. The pathogenicity of the associated genes is classified according to the guidelines of the American College of Medical Genetics and Genomics and the Association of Molecular Pathology [19]. However, many reported variants are interpreted as variants of uncertain significance [20]. In such cases, variant functionality can be tested using co-segregation within the family (Fig. 4) [21].
Candidates for genetic testing usually include adults with LDL-C ≥ 190 mg/dL in the absence of other secondary causes, children or adolescents with LDL-C ≥ 160 mg/dL and a family history of premature CAD or severe hypercholesterolemia. As the cost is partly covered by the National Health Insurance Service (NHIS), further awareness and information on FH in health professionals seems to increase the clinical and genetic diagnosis rate of the patients.
Differential diagnosis
Other secondary and primary hypercholesterolemia should also be ruled out. Secondary etiologies include hypothyroidism, nephrotic syndrome, cholestasis, acute intermittent porphyria, and pharmacological agents (e.g., thiazide and cyclosporine) [22]. The primary etiologies include sitosterolemia and familial combined hyperlipidemia. Sitosterolemia, a rare genetic disease, is accompanied by high levels of circulating plant sterols and is caused by loss-of-function mutations in ABCG5/8. This occurs in 1/200,000 people. LDL-C levels can vary but are very high in some affected individuals, such as children, and can be influenced by diet. Although confirmative diagnosis requires the identification of pathogenic mutations, lack of facility for sitosterol assay and genetic testing can be an obstacle [23]. Individuals with familial combined hyperlipidemia show high very-low-density lipoprotein and LDL-C levels and low HDL-C levels. The prevalence is 1/100 to 200. Familial hyperlipidemia is an important cause of premature CAD. Although the molecular background is uncertain, a polygenic cause is a plausible mechanism, and secondary factors can also affect the phenotype. Patients typically have total cholesterol of 200 to 400 mg/dL, triglyceride of 200 to 600 mg/dL, HDL-C of < 40 to 50 mg/dL and family history. To date, there is no standard definition of this disease, but the most common clues are simultaneous elevation of apoB (≥ 120 mg/dL) and triglycerides (≥ 133 mg/dL) [24].
SCREENING OF FH
The prevalence of FH may be higher than that in previous reports in many countries because of underdiagnosis due to a lack of awareness of its clinical significance. Guidelines and consensus statements on FH referred to the importance of screening, especially cascade screening, for early diagnosis of FH [6,17,25,26]. In addition, they suggested FH screening in accordance with country-specific situations and consensus. Assaying LDL-C levels is crucial for the early detection and treatment of FH, as patients with this disease are usually asymptomatic until atherosclerotic cardiovascular disease (ASCVD) occurs. The 2018 Korean guidelines on dyslipidemia recommended universal screening of dyslipidemia in adults aged ≥ 21 years and younger with other risk factors every 4 to 6 years (i.e., family history of cardiovascular disease and severe dyslipidemia) [27]. However, if a person has a family history of cardiovascular risk factors, lipid tests can be performed at any age.
FH needs to be considered in individuals with CAD aged < 55 years for men and < 60 years for women, or severe elevation of LDL-C (≥ 190 mg/dL in adults and ≥ 150 mg/dL in children), or tendon xanthomas in themselves or relatives, or family history of premature cardiovascular disease [5,27]. Individuals with skin xanthoma, xanthelasma, or premature arcus cornealis should also be considered for FH screening. However, many patients with FH take lipid-lowering agents before they are suspected of having FH. Therefore, the detection of patients tends to be delayed. In this regard, it is critical to improve awareness of FH among health professionals to check the clinical findings and family history compatible with this disease.
Cascade screening of family members of index cases is well known as the most efficient and cost-effective approach for the early detection and treatment of new FH. Cascade screening includes lipid profiles and genetic testing of the first-degree relatives of an index case. Well-organized screening programs can improve FH outcomes, and many countries are introducing programs adapted to each country. Universal screening is another way to detect patients with FH and is undertaken in some countries [13].
TREATMENT OF FH
It is critical to initiate LLT as soon as possible when FH is diagnosed. The simultaneous control of other cardiovascular risk factors is essential because the main purpose of FH treatment is to prevent ASCVD. Non-pharmacological therapies, such as therapeutic diet modification and exercise are largely similar to other guidelines for dyslipidemia [13,27]. Briefly, total fat, saturated fat, trans-fatty acid, cholesterol, carbohydrate, sugar, and alcohol intake needs to be limited, and their limits are suggested. Conversely, the active intake of fiber-rich food, whole- and multigrain, vegetables, fish, and fresh fruits is recommended. Regular performance of aerobic and resistance exercises is recommended [27]. Moderate-intensity aerobic exercise at least 30 minutes for four to six times a week and regular resistance exercise at least twice a week are recommended.
The first-line pharmacotherapy is statins, which are usually administered at a high-intensity. Many patients may not reach the LDL-C target with statin monotherapy, and ezetimibe can be added as a second-line agent. PCSK9 inhibitors can be added if patients do not achieve the target with a maximal tolerable dose of statin plus ezetimibe (Table 3) [5,15,28,29]. A study supported by the Korean Society of Lipid and Atherosclerosis found that achievement rates of LDL-C < 100 mg/dL or 50% reduction of LDL-C by the maximal statin/ezetimibe combination were not high in Koreans with FH [30]. Notably, the response to LLT can be affected by an individual’s genotype associated with FH [31].
Statin
Most international guidelines currently recommend maximal tolerable dose statins as the first-line therapy in patients with FH (Table 3) [5,29]. No randomized clinical trials have investigated the effects of statins in patients with FH. However, a cohort study from the Netherlands showed a 76% lower risk of coronary heart disease in patients with FH who received statin therapy at lower dose than the current guidelines [32]. In addition, a retrospective study from the Netherlands demonstrated a 50% reduction in CAD occurrence and mortality with moderate- to high-intensity statin therapy [33]. Furthermore, meta-analysis provides evidence of the benefits of intensive statin therapy [34].
Ezetimibe
The most recent guidelines recommend this agent as a second-line drug for LLT (Table 3). Ezetimibe, a cholesterol absorption inhibitor, reduces cardiovascular events when combined with a moderate-intensity statin [35]. Another study showed that a combination of statin and ezetimibe induced plaque regression compared to statin monotherapy [36]. Statin/ezetimibe combination results in strong LDL-C reduction (≥ 50% from the baseline) and is relatively safe tool of LLT [37].
PCSK9 inhibitor-monoclonal antibodies
These agents are recommended in patients with FH and are very high-risk when the LDL-C target is not achieved after the use of maximal tolerable dose statin/ezetimibe (Table 3). The results of the Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk (FOURIER) [38] and Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab (ODYSSEY-OUTCOMES) [39] studies showed the cardiovascular benefits of evolocumab and alirocumab, providing a scientific basis for anti-PCSK9 antibody use in patients with FH. The percentage reduction in LDL-C by these agents is not smaller than that in patients without FH [5]. These agents are also considered for patients who experience statin intolerance. However, it is more expensive than other oral agents, and it is difficult but important to determine appropriate LDL-C levels to start anti-PCSK9 antibodies in specific risk groups with consideration of cost-effectiveness [29,40,41].
Bile acid-binding resin
The addition of this agent can be considered for severe hypercholesterolemia (Table 3). Although bile acid-binding resin has an LDL-C-lowering effect, its use is limited as there are no clinical outcome studies.
Other and emerging treatment
Mipomersen is an oligonucleotide derivative that binds apoB mRNA and inhibits the production of all apoB-containing lipoproteins. Its action is independent of LDLR expression and has been developed as an adjunct agent, particularly for patients with homozygous FH (HoFH). In these patients, mipomersen reduced LDL-C levels by 21%. Adverse events include injection site reactions, liver enzyme elevation, and increased hepatic fat content [42]. However, it is not currently available in Korea. Lomitapide, a microsomal triglyceride transfer protein inhibitor, reduces the assembly and secretion of apoB-containing lipoproteins in Golgi complex. Lomitapide reduces LDL-C by 38% to 50% in patients with HoFH [43]. It lowers LDL-C levels in an LDLR-independent manner and leads to the accumulation of hepatic triglycerides and hepatiosteatosis, and liver enzyme elevation. However, this agent is not available in Korea. Inclisiran, a synthetic small interfering RNA, inhibits PCSK9 synthesis and is highly effective at lowering LDL-C levels. In a clinical trial in HeFH, it reduced LDL-C by 48% compared to placebo, with a similar rate of adverse events [44]. A long dosing interval is expected to increase patient adherence. However, this agent is not yet available in Korea. Angiopoietin-like 3 (ANGPTL3) is a protein that inhibits lipoprotein- and endothelial lipase. Evinacumab is an antibody against ANGPTL3 that reduced LDL-C and triglyceride levels by up to 56% in patients with HeFH [45] and HoFH [46] in phase 2 and 3 clinical trials, respectively.
Lipoprotein apheresis is the method remove lipoproteins from the blood and it is important for patients whose lipid-lowering response is not sufficient to pharmacological agents. It has been occasionally used in patients with HoFH or severe HeFH. When applied, LDL-C decreases by 50% to 70%. However, owing to its invasiveness, the patient’s quality of life can be negatively affected [5].
LDL-C targets
Ideal targets are < 55 mg/dL in patients with ASCVD or major risk factors and < 70 mg/dL in those without ASCVD or risk factors [5]. However, achieving these targets is difficult in many cases even after 3-drug combination therapy [47], 50% reduction in LDL-C levels and < 70 mg/dL in patients with ASCVD or major risk factors and < 100 mg/dL in those without these conditions are recommended as realistic second best targets in many other guidelines [6,28,48]. In a study supported by the Korean Society of Lipid and Atherosclerosis analyzing individuals with severe hypercholesterolemia, those who achieved LDL-C < 100 mg/dL after statin therapy showed lower cardiovascular risk than those who did not [8].
HOMOZYGOUS FH
HoFH is a rare, but life-threatening disease. The prevalence of HoFH is approximately 1/1 million. However, it is reported to be 1/160,000 to 300,000 in a recent report [9]. Because the exposure of vessels of patients with HoFH to lipids is more severe than that of patients with HeFH, the incidence of CAD under the age of 20 is not uncommon.
The clinical picture is characterized by extensive xanthomas, marked premature and progressive cardiovascular disease, and untreated LDL-C level > 500 mg/dL or treated LDL-C level ≥ 300 mg/dL. Many patients develop CAD and aortic stenosis before the age of 20 years and can die before 30 years. In children, early symptoms and signs are often linked to aortic stenosis and regurgitation due to the massive accumulation of cholesterol at the valves [25].
Diagnosis
The most well known diagnostic criteria for HoFH are from the consensus panel on FH of the European Atherosclerosis Society in 2014 [25]. It included DNA mutations, LDL-C levels, physical findings, and family history (Table 4). However, strict application of genetic diagnostic criteira can affect insurance benefits of pharmacological agents. Genetic analysis is considered to confirm the clinical diagnosis, facilitate testing of family members, and assist diagnosis where the clinical presentation is borderline between those of HoFH and HeFH [25,49].
Monitoring
Patients with suspected diagnoses need to be referred to a specialist for comprehensive management. Regular screening for CAD or aortic disease is recommended. Patients should undergo cardiovascular evaluation at diagnosis, with subsequent annual echocardiographic evaluation, stress testing, and, if available, computed tomography coronary angiography every 5 years. Emergency education is needed, and clinical assessment is recommended every 6 months.
Treatment
Early detection of HoFH during childhood is crucial. The aim of treatment in HoFH is to start LLT as early as possible and obtain cholesterol levels as low as possible. The treatment targets are 100, 135, and 70 mg/dL in adults, children, and patients with ASCVD, respectively. Lifestyle modification, statin/ezetimibe, and lipoprotein apheresis (if available) are essential for treatment. Lipoprotein apheresis is recommended at the age of 5 years or at least 8 years. New therapeutics such as PCSK9 inhibitors, lomitapide, or mipomersen can be added. The Korean Food and Drug Administration approved PCSK9 inhibitors for HoFH, whereas mipomersen and lomitapide are not yet available in Korea. Evinacumab, an anti-ANGPTL3 antibody, has been approved in some countries for the treatment of HoFH. The control of other cardiovascular risk factors is important and aspirin needs to be considered.
FH IN SPECIAL POPULATIONS
Children
Diagnosis
In a child, FH is diagnosed based on phenotypic criteria, including elevated LDL-C plus a family history of premature CAD and/or elevated LDL-C or positive genetic testing [50]. In children with the above-mentioned family history, the accepted cut-off is ≥ 160 mg/dL. If a parent has a known genetic defect, the child’s diagnostic level of LDL-C can be lower. A child suspected of having FH needs to be screened from the age of 5 years. If possible, genetic testing is recommended for a child when a pathogenic mutation is identified in the family. As cholesterol levels may change during puberty, repeated tests should be performed after this period to confirm FH. Screening for HoFH in children should be undertaken as early as possible.
Treatment
Children with FH should be educated to adopt a proper diet and be treated with statins from 8 to 10 years old. The child’s phenotype, particularly the LDL-C level, is considered when starting pharmacotherapy. The LDL-C target level is < 135 mg/dL [5]. Statin treatment should be initiated at low doses and increased to reach the target. In 2021, the United States Food and Drug Administration approved evolocumab [51], an anti-PCSK9 antibody, for the add-on treatment of children with FH older than 10 years.
Pregnancy
Contraception and pregnancy are key issues in women with FH and should be appropriately discussed. Hormonal contraception is generally contraindicated, and other contraceptive methods are preferred. Women wishing to become pregnant need to be counseled and undergo cardiovascular assessments.
Lifestyle modifications for lipid control are recommended. Although some recent data have denied concerns regarding the teratogenicity of statins, minor effects on the fetal brain maturation cannot be ruled out [52]. Ongoing lipid-lowering drugs need be discontinued one to three months before conception or breastfeeding [6]. For women with severe FH, bile acid-binding resin and/or LDL apheresis can be considered [5]. However, there are no officially recommended LDL-C levels for starting drugs or targets.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





