A Case of Budd - Chiari Syndrome with High Antiphospholipid Antibody in a Patient with Systemic Lupus Erythematosus
Article information
Abstract
Antiphospholipid syndrome is characterized by recurrent episodes of arterial and venous thrombosis, spontaneous fetal losses, thrombocytopenia and persistently elevated levels of Antiphospholipid antibodies. We experienced a case of Budd-Chiari syndrome in a 32-year old female lupus patient who was presented with left leg edema, ascites and esophageal varix. The clinical and laboratory findings were compatible with the cirteria for systemic lupus erythematosus (SLE) and she was found to have anticardiolipin antibody, thrombocytopenia and prolonged partial thromboplastin time. Initially, she was treated with intravenous heparin and urokinase and she was followed up with warfarin, baby aspirin and steroids.
INTRODUCTION
Systemic lupus erythematous (SLE) is an autoimmune disease presenting many kinds of autoantibodies in serum. In case of elevated levels of Antiphospholipid antibodies, the patient presents recurrent episodes of arterial and venous thrombosis, thrombocytopenia and spontaneous fetal losses. This clinical spectrum is termed Antiphospholipid syndrome(APS) in SLE. The vascular thrombosis may occur at any small and large vessels resulting in various clinical manifestations. Here, we reported a case of secondary Antiphospholipid syndrome in a lupus patient who was presented with Budd-Chiari syndrome, due to the thrombosis of the intrahepatic portion of the inferior vena cava (IVC) and persistent elevation of anticardiolipin antibody.
CASE REPORT
A 32-year old Korean female was admitted with a complaint of edema and pain on the left leg for 2 months. In January 1993, she was diagnosed as SLE at another hospital and manifested gross hematuria, proteinuria, azotemia, anemia, thrombocytopenia, hypocomplementemia, positive FANA, positive anti-ds DNA antibody and positive anti-ENA antibody. In May 1993, she was transferred to Kangnam St. Mary’s Hospital with steroid (prednisolone 1mg/kg) medication. We checked the titer of anticardiolipin antibody (ACA), which was 100 GPL IU/ml and followed up the titer of ACA regularly at 2 months’ intervals and the titer of ACA IgG was persistently elevated. Her parity was 0-0-0-0 and she showed thrombocytopenia (35,000/mm3) and prolonged partial thromboplastin time (59.2 sec, control 27.0 sec). Her clinical features were Antiphospholipid syndrome in SLE and she was treated with baby aspirin 100 mg/day and steroid (prednisolone 1mg/kg) at the outpatient clinic. From April 1994, she suffered from edema and pain on the left leg and was admitted to our hospital in June 1994. On admission, she had polyarthralgia but did not complain of fever, malar rash, photosensitivity, oral ulcer, Raynaud’s phenomenon, xerostomia, xerophalmia and allopecia. On physical examination, her blood pressure was 120/80 mmHg. pulse rate 76 beats per minute, respiration rate 20 per minute and body temperature 36.5°C. There were no pathologic lesions in her eyes, ears, nasal or oral mucosa. Chest auscultation and abdomen palpation revealed no abnormalities and peripheral arterial pulsation was normal. There were no cutaneous vasculitis and abnormal neurologic signs.
On laboratory findings, hemoglobin was 10.6 g/dl, hematocrit 32%, white blood cell 8.1×103/mm3 (neutrophil 82%. lymphocyte 10%) and platelet 122×103/mm3. Renal function showed blood urea nitorgen 5.3 mg/dl, creatinine 0.9 mg/dl and 24hr urine protein 6.18 g/day. Urinanalysis showed 0 to 2 white cells and 10 to 20 red cells per high power fields. Lipid profile revealed total cholesterol 170 mg/dl. triglyceride 98 mg/dl and HDL-cholesterol 51 mg/dl. The AST was 16 IU/L. ALT 12 IU/L, alkaline phosphatase 229 IU/L, total bilirubin 0.7 mg/dl, total protein 4.1 g/dl and albumin 2.1 g/dl. The prothrombin time was 11.8 sec (control 12.1 sec) and activated partial thromboplastin time 59.2 sec (control 26.6 sec). On immunologic studies, FANA was positive (homogenous pattern, titer 1 : 1280), anti-ds DNA antibody 5 IU/ml, C3 21 mg/dl and C4 15 mg/dl. Rheumatoid factor was negative and ANCA was positive (GS-ANA, titer 1 : 80). Anti-cardiolipin antibody Ig G was 100 GPL IU/ml. Lupus anticoagulant was positive by the Kaolin clotting test. Anti-ENA and anti-Ro antibodies were all negative. The direct and indirect Coombs’ tests were all negative. The erythrocyte sedimentation rate was 18 mm/hr and C-reactive protein 2.4 mg/l. The immunoglobulin G, A, M levels revelaed 928, 274, 106 mg/dl, respectively. The serum viral hepatitis markers revealed that HBs antigen was negative, HBs antibody positive and HCV antibody negative. Gastrofiberscope showed esophageal varix, grade 2, and gastric fundal varix. Abdominal ultrasonography showed a moderate amount of ascites, moderate splenomegaly and marked coarse increased liver echogenecity. At computed tomography of the abdomen, we could not trace the inferior vena cava at the intrahepatic portion(Fig. 1). Doppler ultrasonography of the left leg showed no thrombosis in the superficial femoral vein and popliteal vein. Inferior and superior venocavograms showed obstructions at the intrahepatic portion of IVC and at both subclavian veins and abnormal collateral vessels were found around the obstructions (Fig. 2, 3). We injected Heparin 5,000 units and Urokinase 500,000 units intravenously at the bolus during venocavogram and further thrombolytic therapy (Heparin 5,000 units/day and Urokinase 500,000 units/day continuously all day long) was done for additional two days. We followed the venocavogram to evaluate the extent of thrombosis, compared with pre-thrombolytic therapy, but we could not find any interval change. We decided to give the patient Warfarin 5 mg/day, Prednisolone 1 mg/kg and baby aspirin 100 mg/day and to follow her up at the outpatient clinic.

Computed tomography. The liver is diffusely enlarged. Following infusion of contrast media, the IVC from the intrahepatic portion to the level of the right renal vein is not opacified. Multiple collateral vessels are noted in the paraaortic region. Both ascedning lumbar veins and azygos-hemiazygos system are prominent. The spleen is also enlarged.
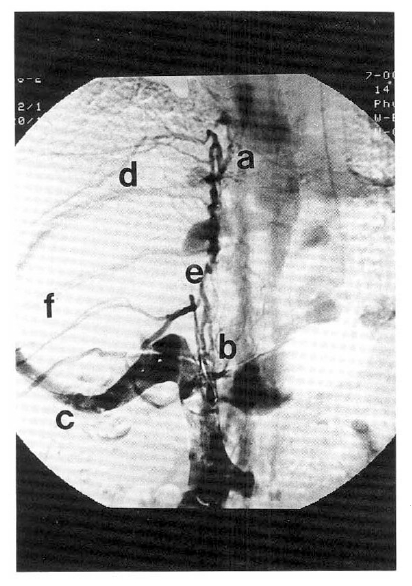
Inferior venocavogram.
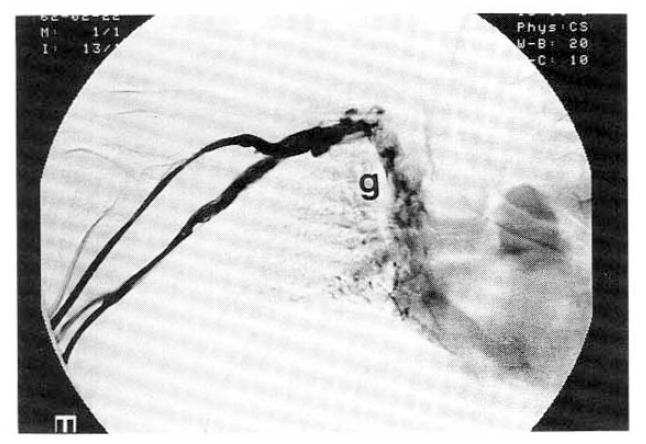
Superior venocavogram.
SVC and IVC venograms show complete obstruction of the intrahepatic portion of IVC (from a to b) with collateral circulation from right inferior hepatic vein (c) to superior hepatic vein (d). Also show collateral channels of azygos and hemiazygos (e) and intercostal veins (f). Left subclavian vein is also obstructed (g).
DISCUSSION
The Antiphospholipid syndrome (APS) is characterized by the production of autoantibodies directed against negatively charged phospholipid. Clinically, it is associated with multiple thrombotic events, thrombocytopenia and recurrent fetal loss2,3). The syndrome can be primary without other systemic disease or secondary, i.e. part of systemic lupus erythematosus4). Antiphospholipid antibodies (aPLs) are also found in certain malignancies5), or as a side effect to certain drugs6–8). The prevalence of aPLs in SLE is 20% to 50% and approximately one-third of SLE patients may have clinical manifestations of APS. The exact mechanism by which aPLs cause APS is still unkown. Since phospholipid is the essential constituent of cell membrane and coagulation pathway, most efforts to elucidate the mechanism have been based on vascular endothelial cell and platelet function. Antiphospholipid antibodies reduce the release of protacyclin at the vascular endothelial cell and enhance the production of thromboxane at the platelet and interfere with many cascades of the coagulation pathway - factor X-converting complex, prothrombinase complex, antithrombin III, thrombomodulin and protein C and S etc. Protein C and a co-factor, protein S, act together to degrade activated factors V and VIII, which leads to inhibit clotting. Antiphospholipid antibodies also inhibit protein C activation which actually results in thrombosis. Antiphospholipid antibodies require a serum co-factor in order to bind to phospholipid, known as β2-glycoprotein 1 (β2-GP1). β2-glycorpotein 1 inhibits the activation of factor XII and the formation of prothrombinase complex, Here, aPLs disturb the inhibitory effect of β2-GP1 resulting in the hypercoagulable state. These thromboses may occur at any organs, causing thrombophlebitis and gangrene of the limbs, Budd-Chiari syndrome9), Addison’s disease, pulmonary embolism, livedo reticularis10), retinal vessel thrombosis11), stroke1,12), myocardial infarction, renal artery thrombosis, hepatic infarction, chorea, psychosis and avascular necrosis of bones.
Budd-Chiari syndrome is characterized by ascites, hepatomegaly, abdominal pain, varicose veins on the lower legs and collateral vessels at the abdominal wall because of the obstruction of the hepatic vein or the inferior vena cava. The syndrome was described, at first, by Budd and Chiari in 1845 and 1899, respectively, and has been mainly reported in East Asia, South Africa and India13,14). The pathogenesis of vascular obstruction is still unclear but some cases of membranous obstruction of the inferior vena cava (MOVC) have been reported in Korea, and MOVC has been known to have a relationship with oral contraceptive agents16) or type B viral hepatitis18). Recently. Budd-Chiari syndrome, associated with lupus anticoagulant, has been reported in many countries17–20), but Budd-Chiari syndrome, as a manifestation of secondary Antiphospholipid syndrome in SLE, is rare and has not yet been reported in Korea.
The patient who has secondary Antiphospholipid syndrome could die unexpectedly, from either cardiopulmonary arrest or circulatory failure, depending on which organ is predominanlty involved by the extensive occlusions. In this instance, the differential diagnosis is often difficult between lupus vasculitis, thrmobotic thrombocytopenic purpura or disseminated intravascular coagulation. To eleminate aPLs, energetic immunosuppression, e.g., with pulse cyclophosphamide, plasmapheresis or gammaglobulin infusion is sometimes effective, but there is usually a rapid rebound to pre-treatment levels shortly after discontinuation of the therapy. Anti-hypertensives, cholesterol-lowering agents and treatment of active nephritis are needed to remove additional risk factors. It is important to prevent occlusive vascular complications in patients who have not experienced thrombosis. Long term use of aspirin is useful in the prevention of arterial occlusion. For the prophylaxis of venous thermbosis, low-dose subcutaneous Heparin (10,000 to 15,000 units/day) is useful and patients who show resistance to the above doses require higher doses of subcutaneous Heparin(25,000 units/day) or intravenous Heparin(40,000 units/day). In the case of occlusive vascular complications, patients require anticoagulation on a long-term basis, and probably for life, because of the tendency of recurrence. Warfarin is used up to 20 mg/day and INR should be kept a a level of 3 to 4 rather than 2 to 2.5. In the case of using Heparin, anticoagulation should be controlled by the recalcification test (Howell test) not by aPTT because of the interference of the aPLs with the latter. To prevent fetal loss, low dose aspirin (30 to 80 mg/day) has proven useful and safe from the beginning of pregnancy. If the patient is receiving warfarin, because of previous thrombotic manifestation, this must be changed to subcutaneous Heparin, at least in the first trimester, since Warfarin is teratogenic. Thrombocytopenia associated with aPLs is usually mild and does not require intervention.