Hepatocyte Growth Factor/c-Met Signaling in Regulating Urokinase Plasminogen Activator in Human Stomach Cancer: A Potential Therapeutic Target for Human Stomach Cancer
Article information
Abstract
Background
Up-regulation of the hepatocyte growth factor (HGF), its transmembrane tyrosine kinase receptor (c-Met), and urokinase type plasminogen activator (uPA), is associated with the development and metastasis of various types of cancers. However, the mechanisms by which HGF/c-Met signaling mediates cancer progression and metastasis are unclear.
Methods
We investigated the roles of HGF/c-Met in tumor progression and metastasis in NUGC-3 and MKN-28 stomach cancer cell lines.
Results
Treatment with HGF increased c-Met phosphorylation in a dose-dependent manner, as well as increasing cell proliferation. HGF treatment also increased the protein level and the activity of uPA in NUGC-3 and MKN-28 cells. A monoclonal antibody against human uPA receptor (uPAR), mAb 3936, inhibited HGF-mediated tumor cell invasion in a dose-dependent manner. Down-regulation of uPA using uPA-shRNA induced a decrease in in vitro cell invasion in NUGC-3 cells.
Conclusions
These results suggest that NUGC-3 and MKN-28 cells express functional c-Met, which may provide a therapeutic target for interfering with metastases of cancer cells by inhibiting uPA and uPAR-mediated proteolysis.
INTRODUCTION
Gastric cancer is one of the most common malignancies in many countries, including Korea. In spite of radical surgery, major obstacles such as local invasion and metastasis result in a very low cure rate. The process of cancer invasion and metastasis is multi-factorial, involving the destruction of the basement membrane and the extracellular matrix1-3). It has been reported that the development of cancer and the formation of metastases are highly associated with increased expressions of hepatocyte growth factor (HGF), its receptor (c-Met) and the urokinase type plasminogen activator (uPA). HGF, known also as scatter factor (SF), is a pleiotropic polypeptide growth factor with a number of biological activities that include cell scattering, stimulation of cell motility, mitogenesis, morphogenesis, angiogenesis, and cellular invasiveness4, 5).
The c-Met proto-oncogene, whose ligand is HGF/SF, was originally identified in a form in which it had been rearranged by transfection of DNA from a human osteosarcoma cell line treated in vitro with a chemical carcinogen6). Subsequently, it was found to be constitutively activated in a human gastric carcinoma cell line7, 8). uPA is one of the plasminogen activators that converts plasminogen to plasmin, a trypsin-like enzyme with broad specificity. An over-expression of uPA has been reported in many malignant tumors, including lung, breast and colon cancers9-11). Previous studies have shown that antibodies specific for urokinase can inhibit the metastatic dissemination of tumor cells in animal models12). Furthermore, in vitro invasion assays using cultured human tumor cells have demonstrated that uPA activity is essential for the invasive phenotype of these cells3). The uPA activity is temporally and spatially regulated by a specific uPA receptor (uPAR), which has been identified on the surface of monocytes and in many cultured cancer cell lines14). Receptor-bound uPA can activate plasminogen with a higher efficiency than free uPA because of its concentration and localization in the immediate peri-cellular environment15, 16).
In this study, we conducted in vitro experiments to determine the function of HGF/c-Met signaling and its direct effects on the growth and invasion of stomach cancer cell lines. The knowledge gained from this research might provide a therapeutic basis for understanding how the inhibition of uPA and uPAR-mediated proteolysis may hinder the process of invasion and metastasis of tumors.
MATERIALS AND METHODS
Cell cultures
Two human gastric cancer cell lines, the poorly differentiated adenocarcinoma, NUGC-3 and the moderately differentiated tubular adenocarcinoma, MKN-28, were obtained from the Korea Cell Line Bank. Cells were maintained in RPMI 1640 medium (Life technologies Inc., Gaithersburg, MD) containing 10% fetal calf serum (FCS) in an incubator under humidified atmosphere of 5% CO2 /95% air at 37℃.
Protein extraction
NUGC-3 and MKN-28 cells were cultured in DMEM supplemented with 10% FCS and incubated for 24 hours at 37℃ in a humidified atmosphere containing 5% CO2. The cells were serum-starved for 24 hours and then treated with increasing concentrations (0, 10, 40 ng/mL) of HGF for 15 min or over increased time periods (0, 1, 3, 10 or 30 min) with 10 ng/mL HGF. Cells were lysed in a lysis buffer (20 mM Tris-HCl pH 8.0, 137 mM NaCl, 1% Triton X-100, 1 mM Na3VO4, 2 mM EDTA, 1 mM phenymethylsulfonyl fluofide, 20 M leupeptin and 0.15 U/mL aprotinin) and centrifuged at 12,000 g for 5 min at 4C. The protein concentrations in the supernatants were quantified by the bicinchoninic acid (BCA) method (Pierce Biotechnology Inc., Rockford, IL) using bovine serum albumin as a standard.
Immunoprecipitation for functional c-Met protein
Two hundred micrograms of protein were mixed with 1 g of a mouse polyclonal antibody against c-Met (clone DO-24, UBI, Lake Placid, NY) and protein A/G agarose. The reaction mixture was incubated overnight at 4℃ with constant stirring and washed 3 times with a RIPA solution [1% NP40, 0.5% sodium deoxycholate and 0.1% SDS in phosphate buffered saline (PBS)]. The proteins were eluted with an SDS-sample buffer (0.5% Tris, 10% SDS, 1M DTT, glycerol and 1% bromophenol blue) after 5 min of heat treatment at 100℃. The proteins were separated on a 7.5% SDS-polyacrylamide gel, transferred to a nitrocellulose membrane (Amersham Pharmacia Biotech., Piscataway, NJ) and probed with an anti-phosphotyrosine antibody (clone 4G10, diluted 1:5000) (Upstate Biotechnology Inc., Lake Placid, NY), or an anti-c-Met antibody (clone C-28, diluted 1:2000, Santa Cruz Biotechnology, Santa Cruz, CA). Peroxidase-conjugated secondary antibodies were applied and the immunoreactive proteins were visualized with ECL chemiluminescence solution (Amersham Pharmacia Biotech, Piscataway, NJ).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cells (1,000/well) in RPMI media containing 5% FCS were seeded into a 96-well plate and incubated for 24 hours. Following serum-starvation for 24 hours, cells were treated for 72 hours with HGF (40 ng/mL) containing 10% FCS. At the end of the incubation period, 50 L of 2 mg/mL MTT solution was added and incubated for an additional 3 hours at 37℃. The MTT solution was carefully removed by aspiration and the resulting formazan crystals converted by the viable cells were dissolved in 100l dimethyl sulfoxide. Absorbance at 570 nm was measured with a Biorad multiscan plate reader. Cell proliferation was expressed as a percentage of HGF-untreated control cells.
Gel zymography
The uPA protein activity was determined by gel zymography. Equal amounts of proteins were resolved under non-reducing conditions in a 10% SDS-polyacrylamide gel impregnated with 0.4% casein and 15 µg/mL human plasminogen as a substrate. After electrophoresis, gels were washed for two hours in a solution of 50 mM Tris-HCl, pH 7.5, containing 2% triton X-100, and 0.02% NaN3. The gels were then incubated overnight in a solution of 100 mM Tris-HCl, pH 7.5 and 150 mM NaCl. Enzyme activity was determined by the negatively stained regions following staining with 0.2% Coomassie blue solution in methanol:acetic acid: water (4:1:5) and de-staining in the same solution without any dye.
Standard two chamber invasion assay
Cells (1×104) and mAb 3936 (0~25 ng/mL) were placed in the upper chamber of a Matrigel migration chamber with 0.8-micron pores (Fisher Scientific, Houston, TX). Media containing 5% FCS and HGF (0 or 40 ng/mL), with or without mAb 3936 (0~25 ng/mL), were added to the bottom chamber. After incubation for 48 hours, cells were fixed and stained using a HEMA 3 stain set (Curtis Matheson Scientific, Houston, TX) according to the manufacturer's instruction. The stained filter membrane was cut and placed on a glass slide. The migrated cells were then counted under light microscopy (10 fields at 200x power).
uPA knock-down with short hairpin RNA (shRNA)
The human uPA-specific shRNA expression vector (uPA-shRNA, RHS1764-9494319) containing a uPA-targeted siRNA sequence (acctcctctcctccagaagaat) corresponding to nucleotides 588-608 of the coding region was purchased from Open Biosystems (Huntsville, AL). NUGC-3 cells were transfected with uPA-shRNA using Lipofectamine (Life Technologies Inc., Gaithersburg, MD). Clonal selection was conducted by culturing with G418 (300 µg/mL) followed by serial dilution of the cells. Stable transfectant clones with a low expression of the target genes were identified by RT-PCR. The primers for uPA RT-PCR were as follows: sense, 5'-gtggcc aaaagactctgagg-3' (positions 25-44) and antisense, 5'-gccgtaca tgaagcagtgttg-3' (positions 209-190).
Western blot analysis
The proteins (100 g) were separated on a 10% SDSpolyacryamide gel, transferred to a nitrocellulose membrane, probed with anti-phospho ERK (T202/Y204, Cell Signaling) and anti-ERK antibodies (Cell Signaling). uPA secreted into the media and uPAR in HGF-treated cells were analyzed by Western blotting using a rabbit polyclonal antibody against human uPA (389, American Diagnostica, Greenwich, CT) and a mouse monoclonal antibody against human uPAR (3936, American Diagnostica, Greenwich, CT). The protein levels were quantified using a Bio-1D program (Vilber Lourmat, Torcy, France).
Statistical analysis
Values are expressed as means±SD. Student's t-test was employed for the analyses. A p-value of less than 0.05 was considered statistically significant.
RESULTS
HGF-mediated c-Met phosphorylation
In order to determine whether NUGC-3 and MKN-28 cells harbor functional c-Met protein, cells were treated with or without HGF and c-Met phosphorylation was measured by immunoprecipitation. Treatment with HGF increased c-Met phosphorylation in a dose-dependent manner (Figure 1A). HGF-mediated c-Met phosphorylation occurred within 1-3 min, and gradually declined by 30 min (Figure 2B). These findings suggest that NUGC-3 and MKN-28 cells have functional c-met protein, and that HGF exhibits a great capacity to auto-phosphorylate c-Met in both cell lines.
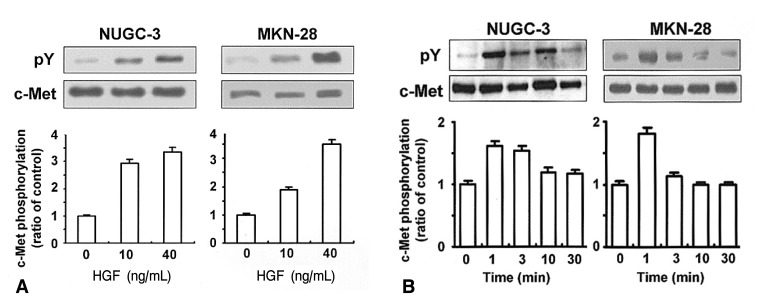
Effects of HGF on c-Met phosphorylation. Cells were cultured in RPMI 1640 media containing 10% FCS, and serum-starved for 24 h. Cells were stimulated with increasing concentrations of HGF (0, 10, 40 ng/mL) for 15 min (A), or with increasing exposure time with 10 ng/mL HGF (B). Following protein extraction, c-Met protein was immunoprecipitated with a c-Met antibody and HGF-mediated c-Met phosphorylation was analyzed by Western blotting with a phosphotyrosine (pY) antibody. The levels of c-Met phosphorylation were normalized for the levels of c-Met protein. Values are means SD of triplicates of three independent experiments.

Effects of HGF on cell proliferation. Cells (1,000/well) were seeded in 96 well plates with RPMI 1640 media supplemented with 5% FCS and incubated for 24 h. After serum-starvation for 24 h, cells were treated for 72 h with or without HGF (0 or 40 ng/mL). Cell proliferation was measured by MTT assays and expressed as a percentage of HGF-untreated control cells. Values are means SD of triplicates of three independent experiments and statistical significance was estimated by Student's t-test (*, p<0.05).
Effect of HGF on cell proliferation
To determine whether HGF might affect cell proliferation in NUGC-3 and MKN-28 cells, HGF-stimulated cell proliferation was measured by MTT assay. HGF treatment produced a greater than 20~70% increase in cell growth, compared to untreated control cells (Figure 2).
Induction of uPA and uPAR following treatment with HGF
Since the addition of exogenous HGF can strongly stimulate an invasive metastatic phenotype, we were interested in determining the downstream effects of this phenotype, focusing primarily on the proteolysis network. The protein level of uPA secreted into the media was 3 fold higher in the HGF-treated NUGC-3 cells than in the HGF-untreated NUGC-3 cells (Figure 3A). uPA protein was also detected in MKN-28 cells in a pattern similar to the NUGC-3 cells. The expression level of uPA protein was lower in MKN-28 cells than in NUGC-3 cells. The activity of uPA measured by casein/plasminogen zymography was increased by treatment with HGF in a dose-dependent manner in cultures of both cells (Figure 3B). Protein levels of uPAR were also increased in both cell lines by treatment with HGF (Figure 4). These findings suggest that one of the effects of HGF/c-Met signaling is mediated by increases in protein level, as well as activity levels, of uPA.

Effects of HGF on uPA expression. Cells were cultured in RPMI 1640 media supplemented with 10% FCS, and serum-starved for 24 h. Cells were treated with increasing concentration of HGF (0, 10 or 40 ng/mL) for 48 h. uPA protein levels and uPA activity secreted into the media were analyzed by Western blotting (A) and casein/plasminogen zymography (B). Values are means SD of triplicates of three independent experiments; statistical significance was estimated by Student's t-test (*, p<0.05; **, p<0.01).

The effect of HGF on uPAR expression. Cell were cultured in RPMI-10% FCS, washed and incubated for an additional 24 h serum free medium. Cells were then stimulated with HGF 0, 10, 40 ng/mL for 48 h. The conditioned medium was harvested and aliquots of conditioned medium were analyzed for uPAR using Western blotting under non-reducing conditions.
Effects of HGF and uPAR mAb 3936 on cell invasion
To examine the effects of HGF/c-Met mediated uPA induction on the invasive properties of tumor cell phenotypes, we performed an in vitro invasion assay using a Matrigel migration chamber. The invasiveness of HGF-treated cells was 3.5 fold higher in NUGC-3 cells, and 1.7 fold higher in the MKN-28 cells than in untreated cells. We hypothesized that HGF-mediated uPA up-regulation is responsible for the invasive properties of tumor cell phenotypes, and that the blocking of uPA activity could be a potential target for inhibiting tumor cell invasion. To test this hypothesis, we studied the effects of a uPAR antibody, mAb 3936, on tumor cell invasiveness. mAb 3936-treated cells showed a substantial reduction in their invasive properties in a dose-dependent manner, compared to IgG2a isotype-matched antibody-treated cells (Figure 5). The inhibitory effects of HGF and mAb 3936 on cell invasion were observed to be greater on NUGC-3 cells than on MKN-28 cells. These results strongly suggest that HGF-mediated uPA induction plays an important role in tumor cell invasion. The fact that the invasive properties of these cells can be inhibited by mAb 3936 suggests that regulation of uPA may be a useful therapeutic target for halting metastasis in stomach cancers.
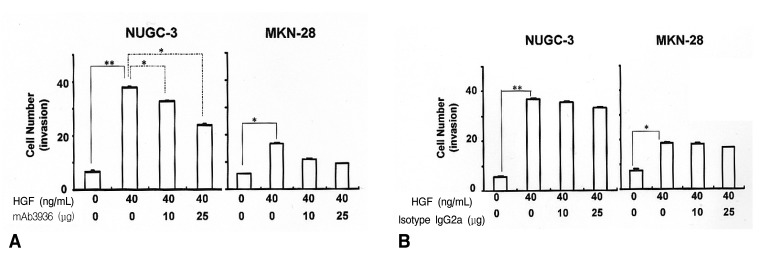
Effects of HGF and mAb 3936 on in vitro invasiveness. Cells in RPMI 1640 media supplemented with 5% FBS were placed in the upper section of Matrigel chambers and treated with or without mAb 3936. The bottom of the chamber was filled with media containing 5% FCS and HGF with or without mAb 3936 (A). An isotype- matched IgG2a antibody was used as a control (B). After 48 h incubation, cells that had migrated through the filter were counted under light microscopy (10 fields at 200x power). Values are means SD of triplicates of three independent experiments. Statistical significance was estimated by Student's t-test (*, p<0.05; **, p<0.01).
Effect of uPA knock-down on cell invasion
To assess the role of uPA in HGF/c-Met mediated cell invasion, we generated uPA-shRNA stable cells. Following selection, cloning and amplification of stable cells, an in vitro invasion using a Matrigel migration chamber assay was measured in the uPA-shRNA cells and the vector-transfected cells. Knock-down of uPA in uPA-shRNA cells was confirmed by RT-PCR (Figure 6A). uPA-shRNA cells showed a decrease in HGF-mediated cell invasion compared to the vector cells (Figure 6B), suggesting that uPA may play an important role in HGF-mediated cell invasion in NUGC-3 cells.
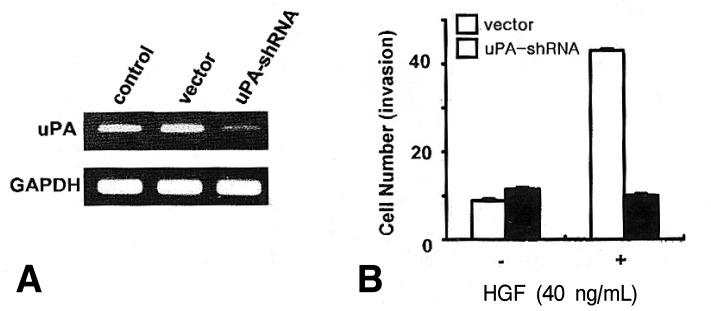
Effects of uPA knock-down on HGF-mediated cell invasiveness in NUGC-3 cells. uPA-shRNA stable cells were generated by transfection of a human uPA-specific shRNA expression vector. Following selection of uPA-shRNA stable cells, knock-down of uPA was confirmed by RT-PCR (A) and in vitro cell invasion was measured by a Matrigel chamber assay (B). Values are means SD of triplicates of three independent experiments.
Activation of ERK following HGF treatment
To elucidate the involvement of MAPK activity in HGF-mediated cell growth, we analyzed ERK phosphorylation in cells treated with HGF. ERK phosphorylation was increased in the HGF-treated cells in a dose-dependent manner (Figure 7).
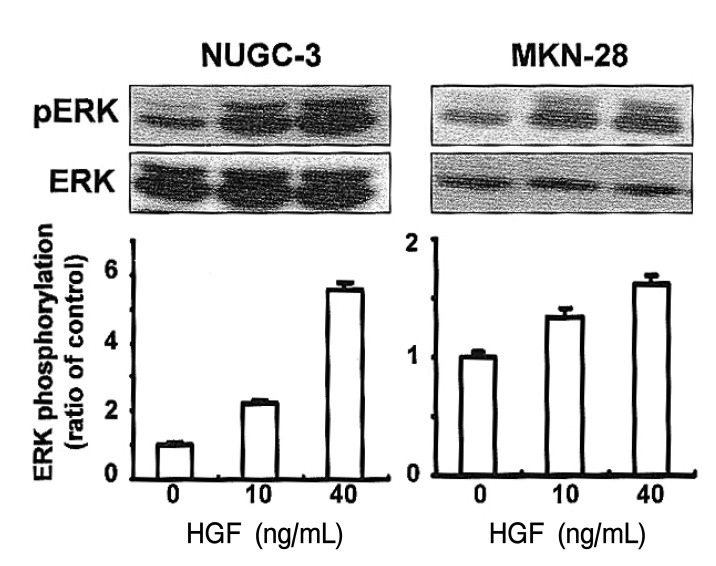
Effects of HGF on ERK1/2 activation. Cell were cultured in RPMI 1640 media supplemented with 10% FCS, and then serum-starved for an additional 24 h. Cells were then stimulated with increasing concentrations of HGF (0, 10, 40 ng/mL) for 15 min. ERK1/2 phosphorylation was analyzed by Western blotting using phophorylated ERK and ERK antibodies. Values are means SD of triplicates of three independent experiments.
DISCUSSION
HGF has been identified as a potent growth factor that stimulates the proliferation of hepatocytes in primary culture17, 18). HGF/SF and its receptor (HGFR) are involved in a variety of normal developmental and homeostatic processes of the body. However, up-regulation of HGF/SF was found to enhance the tumorigenicity of rat bladder carcinoma (NBT-11) and the anchorage-independent growth and tumorigenicity of mouse liver epithelial cell lines20, 21). The expression of transfected HGF/SF in mouse or human tumor cell lines has been reported to promote metastasis to the lung22). Cellular responses to HGF/SF are mediated by the c-Met, a cell surface receptor, which has intrinsic tyrosine kinase activity. Interaction of HGF/SF with c-Met is considered to be an important autocrine and paracrine regulatory process related to neoplastic growth and progression. The generation of an autocrine HGF/SF-c-Met loop by transfected NIH3T3 cells with the murine c-Met gene induced tumorigenic transformation19). Our results showed that c-Met phosphorylation was increased in NUGC-3 and MKN-28 cells in a dose dependent manner by treatment with HGF.
Cancer invasion and metastasis are multifactorial processes and require the coordinated action of cell-secreted proteolytic enzymes and their inhibitors24). Elevated levels of urokinase-type plasminogen have been implicated in this invasive process. HGF/SF upregulates the production of uPA and matrix metalloproteinases (MMPs)23). uPA is a molecular weight of 52,000 serine protease that is released from cells as an inactive zymogen (pro-uPA); through limited proteolysis, it is converted to the active 2-chain uPA25). A specific receptor, urokinase plasminogen activator receptor (uPAR), binds uPA to cell surfaces, and the concomitant cell surface binding of pro uPA and plasminogen strongly enhances the generation of plasmin. Plasmin is capable of degrading most components of the extracellular matrix either directly or through the activation of some procollagenases. The direct binding of HGF to c-Met can initiate uPA gene transcription. Recent data indicate that HGF can upregulate uPA expression and uPA activity in human sarcoma, prostate and pancreatic cells16, 26, 27). Although HGF has previously been shown to enhance uPA activity in several types of cells, we have shown that HGF-mediated uPA up-regulation plays an important role in cellular invasiveness in stomach cancer cells. uPA activity is regulated not only by its inhibitor, but also by the expression of its receptor27). The binding of uPA to its receptor increases uPA activity and its ability to convert plasminogen to plasmin.
Immuno-histochemical studies on various human solid tumors, including stomach cancer, have shown increased levels of uPA and uPAR in the primary tumor tissues,when compared to normal mucosa28-30). However, the role of uPAR in cell invasion and tissue remodeling has been demonstrated primarily in cultured or transplanted cells. In a previous study, we reported increased levels of uPAR in primary stomach cancer tissue as compared to normal tissue. These findings strongly suggest that the PA system may play an important role in tumor invasion and metastasis. It has been reported that the survival rate of patients with tumors displaying high levels of uPAR expression was significantly lower than for those patients without uPAR expression31).
In vitro invasion by pancreatic cells16) and human glioblastoma cells32) can be modulated with an anti-uPAR antibody that blocks uPA binding to the receptor34). In this study, we have shown that an anti-uPAR antibody, mAb 3936, reduces the invasive properties of NUGC-3 and MKN-28 cells in a dose-dependent manner. The levels of uPA expression by HGF-treatment were higher in NUGC-3 cells than in MKN-28 cells. Differences in the levels of HGF-mediated uPA expression might contribute to differences in invasive properties between the two cell lines, as well as the efficiency of mAb 3936 in the inhibition of invasiveness (Figure 5).
Studies of kidney epithelial cells which over-express activated c-Met have demonstrated that uPA gene expression was activated through the MAP kinase pathway34). We attempted to analyze the activity of ERKs in the human stomach cancer cell lines following treatment with HGF. Our data show that ERK was activated in both cell lines by HGF, suggesting that ERK's activation might be an essential signaling mechanism leading to cell invasion (Figure 3). Therefore, further study will be necessary to investigate the involvement of the MAPK cascade as a signal transduction pathway for regulating tumor growth and metastasis in response to HGF in stomach cancer cells.
Acknowledgements
This work was supported by a grant from the Korean Association of Internal Medicine 2004 and by the Korea Science and Engineering Foundation (KOSEF) through the Aging-associated Vascular Disease Research Center at Yeungnam University (R13-2005-005-01003-0(2005)).