Molecular Mechanism of Insulin Resistance in Obesity and Type 2 Diabetes
Article information
Abstract
Insulin resistance is a major risk factor for developing type 2 diabetes caused by the inability of insulin-target tissues to respond properly to insulin, and contributes to the morbidity of obesity. Insulin action involves a series of signaling cascades initiated by insulin binding to its receptor, eliciting receptor autophosphorylation and activation of the receptor tyrosine kinase, resulting in tyrosine phosphorylation of insulin receptor substrates (IRSs). Phosphorylation of IRSs leads to activation of phosphatidylinositol 3-kinase (PI3K) and, subsequently, to activation of Akt and its downstream mediator AS160, all of which are important steps for stimulating glucose transport induced by insulin. Although the mechanisms underlying insulin resistance are not completely understood in skeletal muscle, it is thought to result, at least in part, from impaired insulin-dependent PI3K activation and downstream signaling. This review focuses on the molecular basis of skeletal muscle insulin resistance in obesity and type 2 diabetes. In addition, the effects of insulin-sensitizing agent treatment and lifestyle intervention of human insulin-resistant subjects on insulin signaling cascade are discussed. Furthermore, the role of Rho-kinase, a newly identified regulator of insulin action in insulin control of metabolism, is addressed.
INTRODUCTION
A fundamental mechanism for the maintenance of glucose homeostasis is the rapid action of insulin to stimulate glucose uptake and metabolism in peripheral tissues. Skeletal muscle is the primary site of glucose disposal in the insulin-stimulated state [1]. Resistance to the actions of insulin in skeletal muscle is a major pathogenic factor in type 2 or type 1 diabetes mellitus [2,3]; it also contributes to the morbidity of obesity, and complicates poorly controlled type 1 (autoimmune) diabetes [2]. The ability of insulin to increase glucose transport in skeletal muscle is elicited by the translocation of glucose transporter 4 (Glut4), the major insulin regulated glucose transporter, from intracellular vesicles to the plasma membrane and transverse tubules [4]. In muscle of type 2 diabetic subjects, the expression of the Glut4 gene is normal, and impaired glucose uptake by insulin action most likely results from altered trafficking or impaired function of Glut4 [5-7]. Because glucose transport in response to other stimuli that use different signaling pathways is normal in muscle of type 2 diabetic subjects [4], the resistance to insulin stimulation may be due to impaired insulin signal transduction [8]. In this review, we mainly summarize the updated information on insulin signaling over the past decade, with particular emphasis on the molecular mechanism of human insulin resistance, and also address the physiological role of the newly identified player of insulin action.
Insulin receptor signaling
Insulin signaling involves a cascade of events initiated by insulin binding to its cell surface receptor, followed by receptor autophosphorylation, and activation of receptor tyrosine kinases, which result in tyrosine phosphorylation of insulin receptor substrates (IRSs) including IRS1, IRS2, IRS3, IRS4, Gab1, and Shc [9,10]. Binding of IRSs to the regulatory subunit of phosphoinositide 3-kinase (PI3K) via Src homology 2 (SH2) domains results in activation of PI3K, which phosphorylates membrane phospholipids and phosphatidylinositol 4,5-bisphosphate (PIP2) on the 3' position. This complex activates the 3-phosphoinositide-dependent protein kinases (PDK-1 and PDK-2) resulting in activation of Akt/protein kinase B (PKB) and atypical protein kinase C λ and ζ, (PKCλ/ζ), each of which are serine/threonine kinases [11,12]. Activated Akt phosphorylates its 160 kDa substrate (AS160), which stimulates the translocation of insulin-mediated Glut4 from intracellular vesicles to the plasma membrane [13]. Moreover, activation of PKCλ/ζ is also involved in the regulation of Glut4 translocation in response to insulin [14,15]. However, the insulin receptor (IR) is also dephosphorylated and inactivated by protein tyrosine phosphatases (PTPs), which comprise an extensive family of proteins that exert negative effects on insulin action and glucose metabolism [16,17]. In addition, phosphatase and tension homologue deleted on chromosome 10 (PTEN), a lipid phosphatase, serves as an important negative modulator for the insulin signaling pathway by hydrolyzing phosphatidylinositol 3,4,5-triphosphate to PIP2, antagonizing the PI3K pathway [18,19]. Thus, the physiological regulation of insulin action is controlled by the balance between phosphorylation and dephosphorylation (Fig. 1). Most importantly, the PI3K pathway is thought to be a key component of the insulin signaling cascade, which is necessary for the metabolic effects of insulin on glucose transport and Glu4 translocation [20,21]. Indeed, insulin-stimulated PI3K activity decreases in skeletal muscle of type 2 diabetic subjects [8,22], providing evidence for a defect in insulin signaling that could contribute to impaired Glut4 translocation and insulin resistance.
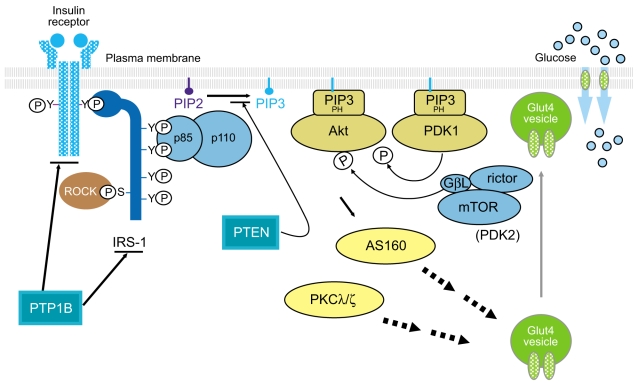
The insulin signaling pathway. PTP1B, protein-tyrosine phosphatase 1B; IRS, insulin receptor substrate; ROCK, Rho-kinase; PIP, phosphatidylinositol phosphate; PTEN, phosphatase and tension homologue deleted on chromosome 10; PH domain, pleckstrin homology domain; PDK, phosphoinositide-dependent protein kinase; GβL, G-protein beta subunit like; mTOR, mammalian target of rapamycin; AS160, 160 kDa Akt substrate; PKCλ/ζ, protein kinase C λ and ζ; Glut4, glucose transporter 4.
Role of Akt in insulin signaling
The serine/threonine kinase Akt is a downstream mediator of PI3K [12]. Three Akt isoforms have been cloned [23,24]; Akt1, Akt2, and Akt3, all of which are ubiquitously expressed in the tissues. Insulin has differential effects on Akt isoforms in a tissue-, isoform-, and species-specific manner [25,26]. In obese rats, insulin-stimulated Akt1 activity decreases in muscle and adipose tissue but increases in liver, while insulin-stimulated Akt2 activity decreases in muscle and liver but increases in adipose tissue [25]. Some studies have shown that inhibition of Akt activation by DN-Akt expression has no significant effect on insulin-stimulated glucose transport, whereas Akt siRNA inhibits insulin-stimulated glucose transport in 3T3-L1 adipocytes [27]. Furthermore, in skeletal muscles from insulin-resistant models, including PTP-1B transgenic mice [28], leukocyte antigen-related phosphatase (LAR) transgenic mice [29], and glucosamine-infused rats [30], insulin-induced Akt activation is normal but insulin-stimulated glucose uptake is impaired. In skeletal muscle of obese insulin resistant humans with or without type 2 diabetes, the effect of insulin on the activity of all Akt isoforms in muscle in vivo is normal [8]. However, at very high insulin levels in vitro, Akt activity is diminished in muscle from non-obese type 2 diabetics [31]. Moreover, Akt2 phosphorylation is impaired in adipocytes from obese type 2 diabetics [32]. Whether these impairments in Akt1 or Akt2 activity are sufficient to play a role in insulin resistant states is unclear. Akt1 knockout mice do not have insulin resistance, although their growth retardation or developmental effects could mask this [33]. Akt2 knockout mice have impaired insulin action on liver and modest effects in muscle and adipocytes that could possibly be secondary to the liver defect [34]. In this regard, our studies have also demonstrated that insulin-stimulated Akt activity in skeletal muscle of these obese rats is very mildly reduced while PI3K activity is markedly reduced, as compared to lean rats [25]. These data lead us to propose the hypothesis that full activation of PI3K is not necessary to maximally activate Akt, and that other pathways that are independent of PI3K are also involved in activation of Akt by insulin, at least in insulin resistant states. Data in support of the hypothesis come from studies showing that insulin-stimulated PI3K activation is reduced by 50% to 60% while Akt activation is normal in hepatoma cells transduced with a dominant-interfering mutant of dynamin [35]. Conceivably, such pathways could be induced in altered metabolic states such as obesity or type 2 diabetes. Thus, the potential impact of altered Akt activity in insulin-dependent glucose metabolism remains uncertain. This important issue can be resolved by studying an animal model that has Akt selectively deleted in peripheral tissues. Until now, there are no data regarding the effects of tissue-specific deletion of Akt on glucose metabolism and insulin signaling.
AS160 is an Akt substrate involved in Glut4 translocation
Investigation of steps down-stream of Akt could shed light on the role of Akt in insulin resistance. Akt directly phosphorylates cytosol proteins that have the RXRXXS/T motif. Using a phosphospecific antibody that only recognizes serine or threonine residues, which are phosphorylated by Akt [36], the Lienhard group has discovered the AS160 from 3T3-L1 adipocytes. This molecule was originally named TBC1 domain family member 4 (TBC1D4) and contains two phosphotyrosine binding (PTB) domains at the NH2 terminus and a Rab GTPase activating protein (GAP) domain at the COOH terminus [37]. In the basal state of 3T3-L1 adipocytes, AS160 was mainly localized in the low density microsomes (LDM) fraction. However, upon insulin stimulation, it was redistributed from the LDM compartment to the cytosol, but these effects were significantly blocked by treatment with a PI3K inhibitor [36]. Given that one of the LDM components are vesicles containing the glucose transporter Glut4 [38], and they move to and fuse with the plasma membrane in response to insulin [39], it is conceivable that AS160 may be involved in the process of GLUT4 trafficking that is dependent on PI3K signaling. The Lienhard group further demonstrated the important role of AS160 in the regulation of Glut4 translocation by showing that insulin causes a marked increase in AS160 phosphorylation at Ser318, Ser570, Ser588, Thr642, and Thr751 residues, and that mutation of these sites inhibits Glut4 translocation in response to insulin [13]. Support for this finding comes from skeletal muscle studies showing that insulin stimulates AS160 phosphorylation in skeletal muscle in a PI3K-dependent fashion, and that contraction and the AMP-activated protein kinase (AMPK) activator aminoimidazole carboxamide ribonucleotide (AICAR) also increases AS160 phosphorylation in isolated rat epitrochlearis muscle and other muscle systems [40-44]. Moreover, in skeletal muscle of human insulin-resistant subjects, including those with polycystic ovary syndrome and type 2 diabetes, the ability of insulin to increase AS160 phosphorylation is significantly impaired [45,46]. Collectively, the current available data provide important evidence that insulin-stimulated phosphorylation of AS160 is required to regulate Glut4 translocation, which is a critical step in controlling glucose homeostasis, and that decreased insulin-induced AS160 phosphorylation in skeletal muscle may play an important role in insulin resistance in vivo.
Role of PKCλ/ζ in the regulation of insulin-mediated glucose transport
The atypical PKC isoforms λ and ζ, are downstream PI3K mediators, and their activation is required for insulin stimulation of glucose uptake [47-49]. Overexpression of a dominant negative mutant of PKCλ or PKCζ abrogates insulin-stimulated glucose transport and Glut4 translocation in adipose [47,50] and muscle cells [48,49]. Overexpression of constitutively active PKCλ in adipocytes [47] or wildtype PKCζ in muscle in vivo [51] enhances both basal and insulin-stimulated glucose transport. In addition, PKCλ and ζ appear to function interchangeably, as overexpression of wild type PKCλ restores the inhibitory effects of a dominant negative mutant of PKCζ on insulin-stimulated Glut4 translocation, and vice versa [52]. The possibility that PKCλ/ζ could play an important role in insulin resistance in vivo is supported by studies showing impaired insulin-stimulated PKCλ/ζ activity in skeletal muscle and adipose tissue of non-obese type 2 diabetic GK rats [53,54]. Moreover, insulin-stimulated PKCλ/ζ activity decreases in cultured myotubes of obese humans with impaired glucose tolerance [55] and in muscle of obese diabetic monkeys [56]. Along with these findings, the Farese group have demonstrated that muscle-specific deletion of PKCλ/ζ causes insulin resistance by reducing insulin-mediated glucose transport in skeletal muscle [15]. Importantly, insulin-stimulated PKCλ/ζ activity is reduced in the muscle of obese nondiabetic and obese type 2 diabetic subjects. This contrasts with our findings that Akt activation is normal in obese and diabetic subjects with similar metabolic characteristics [8]. Thus, although insulin-induced PI3K activity is reduced in type 2 diabetic subjects, not all downstream pathways are similarly affected. These data imply that reduced insulin-stimulated atypical PKC activity may play an important role in insulin resistance in vivo.
Insulin-sensitizer agents and insulin signaling
Thiazolidinediones (TZDs) are a new class of insulin-sensitizing agents being used for the treatment of type 2 diabetes [57]. The molecular targets of these compounds are thought to include the nuclear receptor, peroxisome proliferator activator receptor-γ (PPARγ), which regulates the expression of numerous genes that affect glucose and lipid metabolism [58]. Evidence suggests that TZDs ameliorate insulin resistance in humans primarily by increasing insulin-stimulated glucose disposal in skeletal muscle [59]. Our previous studies revealed that treatment with troglitazone, a member of the TZD family, increases insulin-stimulated IRS-1-associated PI3K activity and Akt activity in skeletal muscle of type 2 diabetic patients [60]. The troglitazone effect on PI3K activity was associated with an increase in the amount of the p110β catalytic subunit of PI3K [60]. Consistently, enhanced Akt phosphorylation was also detected in skeletal muscle in normal, glucose-tolerant, insulin-resistant, first-degree relatives of type 2 diabetic patients [61]. These findings suggest that the mechanism for the insulin-sensitizing effect of TZDs could involve enhanced PI3K activation in skeletal muscle of obese type 2 diabetic subjects. However, Karsson et al. [62] demonstrated that insulin action on Akt phosphorylation and PI3K activity is unaltered in skeletal muscle of human subjects with newly diagnosed type 2 diabetes after treatment of rosiglitazone, another member of the TZD family. This discrepancy could be due to either the nature of the human subjects (obese type 2 diabetic vs. lean type 2 diabetic) or the different kinds of TZDs. From the view of the current human data, it is somewhat unlikely that changes in insulin signaling can fully account for the improvement of insulin sensitivity and glucose disposal in skeletal muscle in response to TZD treatment.
Given that correlative changes in fatty acid metabolism and improvements in glucose homeostasis and insulin sensitivity may imply an indirect effect on skeletal muscle via adipose tissue [63,64], it is possible that some of the effects of TZDs on insulin signaling work through secondary mechanisms. One potential factor is fatty acids, as elevations of fatty acids in plasma and the lipid content of muscle are associated with insulin resistance [65]. As previously reported [66,67], troglitazone treatment tends to lower triglyceride and free fatty acid concentrations. TZDs also reduce accumulation of muscle triglycerides and diacylglycerol [68]. Activation of PKC by elevated diacylglycerol levels in muscle impairs insulin signaling [69,70]. This raises the possibility that decreases in plasma lipid concentrations with TZD treatment could lead to a reduction of diacylglycerol in muscle, reducing PKC activation, resulting in an improvement of the insulin signaling cascade. It is also possible that TZDs reduce intramyocellular lipid content by promoting storage of free fatty acids in adipocyte triglycerides via PPARγ, redirecting free fatty acids from skeletal muscle to adipose tissue. Thus, the ability of troglitazone to improve insulin action in skeletal muscle could involve actions on both adipose tissue and muscle.
Metformin is a member of another class of drugs that are effective in lowering glucose concentrations in patients with type 2 diabetes [71]. A number of studies have demonstrated that metformin inhibits gluconeogenesis, reduces hepatic glucose output, and lowers fasting blood glucose concentration [59,72]. In addition to its effect on the liver, metformin also appears to decrease glucose concentrations by increasing peripheral insulin sensitivity and augmenting insulin-mediated glucose uptake in skeletal muscle of type 2 diabetic subjects [73]. The precise mechanism for this action of metformin is incompletely understood, but in vitro studies indicate it could involve multiple effects, including increased translocation of Glut1 and Glut4 glucose transporters from intracellular vesicles to the cell surface [74] and increased binding of insulin to cell surface insulin receptors [75]. Evidence shows that metformin normalizes insulin receptor tyrosine phosphorylation and PI3K and Akt activity in adipocytes exposed to high insulin levels in vitro for long periods [76]. However, our studies have demonstrated that the effects of metformin on PI3K and Akt activity in skeletal muscle of human insulin resistant subjects with obesity and type 2 diabetes in vivo are unchanged [60]. These data are further confirmed in skeletal muscle of patients with newly diagnosed type 2 diabetes [62]. Overall, it is clear that enhanced insulin-mediated glucose disposal by metformin therapy in type 2 diabetic patients is independent of improved insulin signaling in skeletal muscle of type 2 diabetic subjects.
Effects of weight loss therapy on skeletal muscle insulin signaling
Given that obesity is the major risk factor for developing insulin resistance, it is no doubt that lifestyle intervention that produces weight loss, improves insulin sensitivity. A reduction of fat mass by weight loss results in a significant decrease in lipid oxidation and an enhanced glucose homeostasis [77]. In addition, insulin secretion and plasma insulin concentrations decrease significantly after weight loss. Our work has investigated whether weight loss therapy can alter the ability of insulin to activate PI3K and downstream signaling in skeletal muscle of obese nondiabetic patients. For these studies, ten obese nondiabetic subjects (body mass index [BMI], 30 to 45 kg/m2) were challenged on a very low calorie diet (VLCD) of 600 to 800 calories per day for up to 24 weeks or until 10 to 15% of initial body weight was lost [78]. Once the weight loss goal was met, the subjects were introduced to a weight-maintenance diet. Glucose disposal rate measured by euglycemic hyperinsulinemic clamp was increased by 30% after VLCD treatment, indicating enhanced systemic insulin sensitivity. VLCD treatment significantly increased IRS-1 tyrosine phosphorylation, compared with the pretreatment level [78]. In parallel, insulin-stimulated IRS-1-associated PI3K activity was increased 2-fold post-treatment with VLCD, compared with pretreatment [78]. These changes were independent of the total amount of IRS-1 protein. Importantly, the impaired PKCλ/ζ activation in obese nondiabetic humans was reversed with weight reduction. This could be due to increased signaling upstream of PKCλ/ζ. This, combined with the observation that treating diabetic subjects with TZDs reverses the defect in PKCλ/ζ activity [79], suggests that reversal of the PKCλ/ζ defect might enhance insulin sensitivity in obese insulin-resistant humans. In line with these findings, recent studies of severely obese individuals also demonstrated that weight loss with a very low energy diet (1,883 kJ/day) markedly enhances insulin sensitivity by improving insulin-stimulated glucose disposal [77]. This enhanced sensitivity was accompanied by increased insulin signaling at the level of AS160 and proline-rich Akt substrate 40, which is a component of the mammalian target of the rapamycin nutrient-sensing pathway. Taken together, weight reduction in obese individuals improves insulin sensitivity, which may result from an improvement in PI3K and its downstream signaling in skeletal muscle.
Although our studies have not investigated Glut4 translocation in response to insulin in skeletal muscle of obese subjects that have undergone weight loss, a number of studies have demonstrated that enhancement in the insulin signaling pathway was not accompanied by a significant improvement in Glut4 translocation to the plasma membrane in skeletal muscle from type 2 diabetic patients, TZD-treated type 2 diabetic patients, and obese subjects who underwent gastric bypass surgery [62,80,81]. This could be explained by the fact that intrinsic activity of Glut4 at the plasma membrane is increased or that an unidentified glucose transporter, which may contribute to increased glucose disposal in skeletal muscle, is involved in this event.
Protein tyrosine phosphatase 1B and insulin signaling
Tyrosine phosphorylation of the IR is reversible, and IR dephosphorylation takes place rapidly in intact cells even with the continued presence of insulin [82]. Because a critical regulatory step in insulin signal transduction is the dephosphorylation of signaling molecules by PTPs, it is plausible that enhanced activity of one or more PTPs could lead to insulin resistance. Several studies of obese humans and rodents have reported that the expression and/or activity of specific PTPs, including the transmembrane LAR and the intracellular enzymes protein-tyrosine phosphatase 1B (PTP1B) and Src-homology 2 domain-containing phosphatase-2 (SHP2, SHPTP2, or syp), increase in muscle and adipose tissue [83-87]. LAR and PTP1B show the greatest increases (3-fold in muscle, 2-fold in fat) [83,88]. There is a strong correlation between BMI and total PTP activity toward the IR in both skeletal muscle and adipose tissue from lean and obese subjects [83,88]. Recent studies have also observed increased levels of LAR and PTP1B expression in liver and muscle from obese rodents [89].
Support for the role of PTPs in the regulation of insulin action comes from transgenic and gene knockout studies. The Kahn group generated PTP1B null mice by targeted disruption of the ATG-coding exon [90]. Elchebly et al. [91] targeted exons 5 and 6 (Ex5/6-/-) and also obtained PTP1B-null mice. Both lines of mice have increased insulin sensitivity, manifested by enhanced insulin-stimulated phosphorylation of IR and IRS-1 in muscle and liver. PTP1B-deficient mice have reduced body fat and are protected from diet-induced obesity due, at least in part, to increased basal metabolic rate and total energy expenditure [90]. In addition, insulin-stimulated whole-body glucose disposal is enhanced in PTP1B-deficient mice [90]. Surprisingly, this effect is tissue-specific; insulin-stimulated glucose uptake is elevated in skeletal muscle but not in adipose tissue. These data suggest that overexpression of PTP1B in insulin-target tissues in vivo could contribute to insulin resistance. Consistent with this hypothesis, Zabolotny et al. [28] demonstrated that selective overexpression of PTP1B in skeletal muscle impairs insulin-stimulated PI3K activity and causes mild insulin resistance in vivo. Moreover, recent studies have indicated that liver-specific deletion of PTP1B improves insulin resistance and attenuates diet-induced endoplasmic reticulum stress [92]. Similar to these findings, Haj et al. [93] showed that liver-specific re-expression of PTP1B in PTP1B deficient mice leads to marked attenuation of their enhanced insulin sensitivity. In obese diabetic insulin resistant ob/ob and db/db mice, PTP1B antisense oligonucleotide (ASO) treatment reduces PTP1B protein and mRNA level in liver and fat, and normalizes plasma glucose levels along with improved glucose tolerance and insulin sensitivity [94,95]. Taken together, the current data indicate that inhibition of PTP1B in peripheral tissues may be useful for treating metabolic-related disorders such as obesity and type 2 diabetes. In fact, the development of PTP1B inhibitors has received much attention by the pharmaceutical industry. However, it has been difficult to identify a selective, safe, and effective PTP1B inhibitor, although a novel PTP1B inhibitor (JTT-551) has been suggested as a potential therapeutic agent [96].
Rho-kinase and IRS-1 serine phosphorylation
Rho-kinase (ROCK) is a serine/threonine protein kinase identified as a GTP-Rho-binding protein [97]. There are two isoforms, ROCK1 (also known as ROKβ) [98,99] and ROCK2 (also known as ROKα) [98,100]. ROCK participates in the insulin signaling network by interacting with IRS-1 [101,102]. Our work has demonstrated that inhibiting ROCK decreases insulin-stimulated IRS-1-associated PI3K activity in adipocytes and myotubes. This effect is mainly due to decreased tyrosine phosphorylation of the YXXM motif in IRS-1, which can lead to reduced interaction of IRS-1 with the p85 subunit of PI3K. Indeed, insulin-stimulated IRS-1 binding to the p85 regulatory subunit of PI3K is impaired in adipocytes expressing dominant negative ROCK [103]. By mass spectrometry analysis, we identified the serine residues of IRS-1 at serine 632/635, serine 936, and serine 972, all of which are phosphorylated by ROCK. Interestingly, these sites are close to the YMXM motif domain in IRS-1, which is the binding site of the p85 regulatory subunit of PI3K. Evidence indicates that serine phosphorylation of IRS-1 is a key regulator of insulin signaling [104]. However, the effects of phosphorylation of individual IRS-1 serine residues on insulin signaling appear to be complex and may be context dependent; they are still being defined. Studies of the effects of IRS-1 serine 632/635 phosphorylation on insulin action have yielded conflicting results. Our studies have demonstrated that replacing IRS serines 632 and 635 with alanine causes a significant inhibition of insulin-stimulated IRS-1 tyrosine phosphorylation and PI3K activity [103], suggesting a positive role for IRS-1 serine 632/635 in insulin action.
Our recent studies have demonstrated that global deletion of ROCK1 in mice results in whole-body insulin resistance and impaired skeletal muscle insulin signaling. These effects are independent of changes in body adiposity. Insulin-stimulated IRS-1 serine 632/635 phosphorylation and PI3K activity associated with IRS-1 or phosphotyrosine are impaired in skeletal muscle of ROCK1 deficient mice [105]. Impaired insulin-induced IRS-1 serine 632/635 phosphorylation also accompanies decreased PI3K and ROCK activation in skeletal muscle of obese, insulin-resistant or diabetic mice and rats (unpublished data), suggesting that impaired IRS-1 serine 632/635 phosphorylation may be an important mechanism contributing to the pathogenesis of insulin resistance in obesity. In addition, insulin-stimulated ROCK activity is impaired in skeletal muscle of obese diabetic mice and insulin-resistant humans with obesity and type 2 diabetes (unpublished data). Importantly, insulin-stimulated ROCK1 activity was positively correlated with glucose disposal rate, suggesting that defective ROCK1 activation may contribute to the pathogenesis of human insulin resistance. Thus, our studies identify ROCK1 as a novel player regulating insulin-mediated glucose metabolism in vivo. Further studies of ROCK1 and ROCK2 functions in different metabolic tissues will be needed to precisely delineate ROCK isoform functions that regulate tissue and whole-body insulin sensitivity and glucose homeostasis. The emergence of ROCK1 as an important regulator of insulin action could lead to new treatment approaches for obesity and type 2 diabetes.
CONCLUSION
The earliest defect in the development of type 2 diabetes is insulin resistance characterized by decreased glucose transport and metabolism in skeletal muscle. Studies with the skeletal muscle of type 2 diabetic humans demonstrate impaired insulin activation of the IRS-1/PI3K/Akt signaling pathway, which is a critical step in the regulation of glucose transport in response to insulin. These defects are selectively restored by treatment with an insulin-sensitizing agent and lifestyle changes, representing the core of insulin signaling components. Recent advances have also revealed that insulin action on AS160 phosphorylation is diminished in skeletal muscle of type 2 diabetic patients, and that inhibiting AS160 causes a significant decrease in insulin-dependent translocation of Glut4, suggesting an important role for AS160 in glucose metabolism. Furthermore, defective ROCK activity in skeletal muscle may also contribute to impaired glucose homeostasis in type 2 diabetic patients. A better understanding of the disease pathogenesis and the potential alternative pathways for regulating glucose metabolism could lead to new therapeutic targets for obesity and type 2 diabetes by clarifying intracellular defects in the insulin signaling cascade.
Notes
No potential conflict of interest relevant to this article was reported.