Carbon monoxide: present and future indications for a medical gas
Article information
Abstract
Gaseous molecules continue to hold new promise in molecular medicine as experimental and clinical therapeutics. The low molecular weight gas carbon monoxide (CO), and similar gaseous molecules (e.g., H2S, nitric oxide) have been implicated as potential inhalation therapies in inflammatory diseases. At high concentration, CO represents a toxic inhalation hazard, and is a common component of air pollution. CO is also produced endogenously as a product of heme degradation catalyzed by heme oxygenase enzymes. CO binds avidly to hemoglobin, causing hypoxemia and decreased oxygen delivery to tissues at high concentrations. At physiological concentrations, CO may have endogenous roles as a signal transduction molecule in the regulation of neural and vascular function and cellular homeostasis. CO has been demonstrated to act as an effective anti-inflammatory agent in preclinical animal models of inflammation, acute lung injury, sepsis, ischemia/reperfusion injury, and organ transplantation. Additional experimental indications for this gas include pulmonary fibrosis, pulmonary hypertension, metabolic diseases, and preeclampsia. The development of chemical CO releasing compounds constitutes a novel pharmaceutical approach to CO delivery with demonstrated effectiveness in sepsis models. Current and pending clinical evaluation will determine the usefulness of this gas as a therapeutic in human disease.
INTRODUCTION
Over the last two decades, gaseous molecules have joined the ranks of experimental therapeutics with potential applications in the treatment of pulmonary or systemic diseases. The principle advantage of gases in therapy is in their mode of administration (i.e., inhalation), which is practical for clinical use and noninvasive [1]. Gases which have known medical applications include a number of low molecular weight substances (e.g., H2, O2, N2O, CO2, He, Xe, N2, O3) [2,3]. In addition to these, three gases have gained widespread attention as novel therapeutics with anti-inflammatory and vasoregulatory properties: nitric oxide (NO) [4,5], carbon monoxide (CO) [5-11], and hydrogen sulfide (H2S) (Table 1; for chemical physical properties of these gases) [5,12-16]. In addition to their presence as ubiquitous contaminants of indoor and outdoor air, these three gases share an important similarity in that they are all produced in the body as the natural products of enzymatic reactions (Table 2) [13-15,17-23].
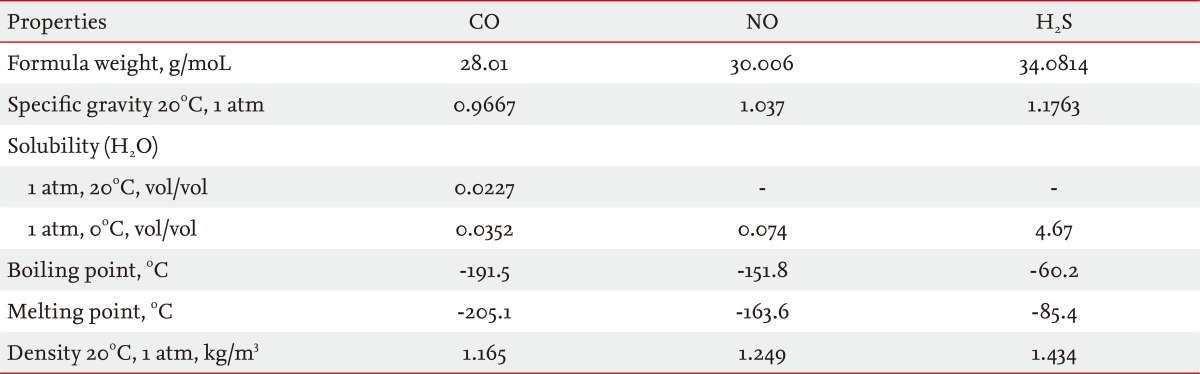
Chemical and physical properties of the medicinal gases
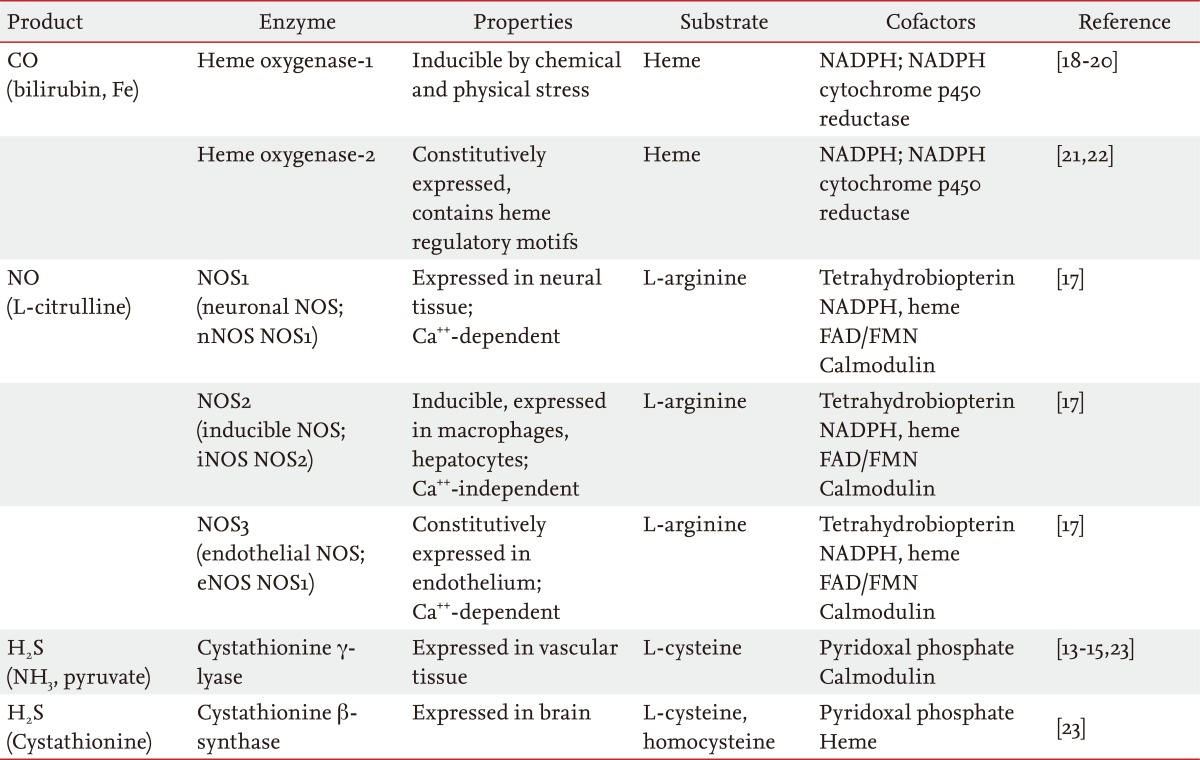
Enzymatic generation of small gaseous mediators
The recognition that gases could exert physiological functions began with the identification of NO as an endogenously-produced regulator of vasodilatation and vascular cell proliferation [24-26]. These effects of NO are mediated by the binding of this gas to the heme moiety of soluble guanylate cyclase (sGC), which stimulates the production of 3',5'-cyclic guanosine monophosphate (cGMP) [27-29]. NO is a reactive free radical that can participate in oxygen and metal-dependent redox reactions, which arises as the product of constitutive and inducible nitric oxide synthase (NOS) enzymes [17]. Inhaled NO, which acts as a selective pulmonary vasodilator [30,31], has been investigated for its therapeutic potential in several diseases including acute respiratory distress syndrome and pulmonary hypertension [31-33].
CO, a diatomic low molecular weight gas, shares similarities with NO in terms of molecular size and structure [34]. CO however, is a relatively stable non-radical gas that typically reacts in biological systems with metal centers of hemoproteins [35]. The endogenous production of CO as the natural product of hemoglobin turnover has been recognized since the middle of the twentieth century [36-38]. However the production of CO in biological systems was previously regarded by the scientific community as a metabolic elimination product. In 1968 to 1969, the heme oxygenase (HO) enzyme system responsible for the catalytic turnover of heme, was characterized and identified as a major source of CO in the body [18,19,39]. The inducible form of this enzyme, HO-1, was identified as identical to the major 32 to 34 kDa stress protein responsive to xenobiotic stress [20,40]. Importantly, further studies established an association between the heme metabolic pathway and the cellular stress response involving HO-1, which contributes to cellular adaptation to toxic environmental challenges [20,41,42]. Subsequent investigations sought to determine the physiological function of endogenously produced CO, as well as its role as a mediator of the cytoprotective properties of HO-1 [7,43-46]. Although many investigations have employed exogenous CO at low concentration, they revealed an impact of this gas on intracellular signaling pathways [43-46]. These studies identified new roles of CO on the regulation of several fundamental biological processes, including vascular tone [47,48], inflammation [43], neurotransmission [44], cell proliferation [49], programmed cell death [45], mitochondrial biogenesis [50], and autophagy [51].
The current prospective status of CO as an inhaled therapeutic is based on extensive preclinical animal testing: reviewed in references [7,11], in models of acute lung injury (ALI) [52-54], ischemia/reperfusion (I/R) injury [55,56], sepsis [57], vascular injury, organ transplantation [58-60]; and others, reviewed in references [5-7,61]. Protective effects of CO have been attributed to several mechanisms, including the regulation of inflammation and innate immune responses, apoptosis, as well as effects on microcirculation and cellular redox balance [5-7,11].
In addition to NO and CO, a third endogenously-produced gaseous molecule H2S has recently emerged as a physiological mediator and candidate therapeutic agent [12-16,62]. H2S which induces a suspended animation-like state in rodents can exert effects on the regulation of vascular tone, inflammation, myocardial contractility, neurotransmission, and insulin secretion [62-66]. H2S has been demonstrated to confer anti-inflammatory properties in several preclinical models, including ALI, and sepsis [67-69].
This review will focus primarily on the therapeutic potential of CO, with an emphasis on mechanistic studies and preclinical animal studies in models of ALI, sepsis, and organ transplantation, as well as its future prospect for clinical use. The pharmacological application of CO in therapy through the use of donor compounds will also be discussed [11]. The reader is referred to other reviews for recent perspectives on the therapeutic potential of NO [4,5], H2S [13,62] and of the other clinically relevant gases [3].
ENVIRONMENTAL AND CLINICAL TOXICOLOGY OF CO EXPOSURE
Environmental CO occurs in the atmosphere from man-made and natural sources, as a by-product of organic combustion. CO is a toxic inhalation hazard, and a common contaminant of indoor and outdoor air [70]. Environmental CO arises primarily as the product of the incomplete oxidation of fossil fuels (e.g., wood, coal, kerosene, and natural gas), and is present at high concentrations in automobile exhaust and tobacco smoke [70].
Indoor levels of CO range from 0.5 to 5 parts per million (ppm) but may reach much higher values (e.g., 100 ppm) with inefficient heating or ventilation, or in the presence of environmental tobacco smoke [71]. In urban areas, ambient levels are typically 20 to 40 ppm, but may peak at much higher levels in heavily congested areas or alongside highways [72].
Inhaled CO diffuses rapidly across alveolar and capillary membranes, with the majority forming a tight binding complex with the oxygen carrier protein hemoglobin to form carboxyhemoglobin (COHb), with a binding affinity for hemoglobin approximately 200 to 250 times that of oxygen [70,73,74]. Partial occupation of CO at the O2 binding sites of hemoglobin inhibits the release of O2 from the remaining heme groups. These effects of CO impair the capacity of the blood to deliver O2, leading to tissue hypoxia [70,73]. The formation of COHb is reversible by removal of the CO source in favor of O2 inspiration. Thus, oxygen therapy is a common antidote for CO poisoning [74,75]. The basal COHb level in man is ~0.1% to 1% in the absence of environmental contamination or smoking. Habitation of heavily populated urban areas with high levels of ambient CO, such as originating from automobile exhaust, or smoking, may increase this background [76,77].
COHb levels of greater than 20% are typically associated with symptoms of clinical toxicity. Acute signs of CO poisoning include dizziness, shortness of breath, and headache. Elevated or chronic exposures may lead to neurotoxicity, cognitive impairment, visual impairment, and unconsciousness, with death occurring in the range of 50% to 80% COHb [74,75]. Recent studies have also identified chronic exposure to elevated ambient CO as a cardiovascular risk factor [78]. The proposed mechanisms for cardiotoxicity associated with chronic low level exposure have been reviewed elsewhere [79].
Although hemoglobin represents the primary systemic target of CO, a direct toxicity of CO has also been described, associated with impaired functioning of cellular hemoprotein targets (e.g., myoglobin, cytochrome c oxidase and cytochrome p450), resulting in the impairment of respiration and other metabolic functions. However, the contribution of these additional cellular and tissue targets to systemic toxicity, usually associated with hypoxemia, remains unclear [74].
THE HO ENZYME SYSTEM: A SOURCE OF BIOLOGICAL CO
The majority of endogenously produced CO (estimated at 85%) arises as the natural product of heme degradation, most of which originates systemically from hemoglobin turnover (Fig. 1) [36-38]. However, the heme pool subject to degradation may also originate from the turnover of other cellular hemoproteins in which it is utilized as cofactor, including cytochrome p450 and other cytochromes [21,80]. Heme is degraded enzymatically by the HOs (EC 1:14:99:3) [18,19,21]. The HO enzymes catalyze the oxidative cleavage of heme at the α-methene bridge carbon atom, to generate biliverdin-IXα, ferrous iron and CO. The HO reaction requires three moles of molecular oxygen for each heme molecule oxidized, and electrons from nicotinamide adenine dinucleotide phosphate (NADPH) cytochrome p450 reductase [19]. Bilirubin-IXα is reduced to biliverdin-IXα by NADPH: biliverdin reductase [81]. Although the HO reaction represents the major enzymatic source of CO, a minor component of endogenous CO may also arise from poorly defined nonheme sources. CO may arise as a by-product of lipid oxidation or as the product of cytochrome p450-dependent metabolism of xenobiotics (e.g., methylene chloride) [82].

Sources of biological carbon monoxide (CO). CO is naturally produced in the human body, primarily as the product of the turnover of hemoglobin and cellular hemoproteins. Heme, which is used as a prosthetic cofactor for hemoproteins, is degraded by the heme oxygenase (HO) (EC 1:14:99:3) enzyme system. HO catalyzes the rate limiting step in heme degradation, to generate biliverdin-IXα, CO, and ferrous iron (Fe II), and requires 3 mol molecular oxygen and nicotinamide adenine dinucleotide phosphate (NADPH). Biliverdin-IXα produced in the HO reaction is reduced to bilirubin-IXα by biliverdin reductase (side chains are labeled as M, methyl; V, vinyl; P, propionate). Nonheme sources may make a minor contribution to exogenous CO production. In the blood, CO binds hemoglobin to form carboxyhemoglobin (COHb). CO may also be inhaled with ambient air, in the context of smoking, accidental or occupational exposure, or as a component of therapy, as discussed in this review.
HOs consist of two major isozymes (HO-1 and HO-2), each the product of distinct genes [21,22,83]. The constitutive isozyme HO-2 is expressed in most tissues, with particular abundance in testis and brain tissue [21]. HO-1, the inducible isozyme, represents a major cellular and tissue homeostatic response to environmental stress signals [20]. HO-1 expression responds to many diverse chemical and physical stimuli, including the natural enzymatic substrate heme, a pro-oxidant compound, oxidants (e.g., H2O2), heavy metals and thiol (SH)-reactive substances, natural antioxidants, deviations in ambient oxygen tension, as well as heat shock (in rodents) [7,20,41,42,84-87]. The induction of HO-1 is mediated primarily by transcriptional regulation [87,88]. The mouse HO-1 (hmox-1) gene 5' regulatory region contains two upstream enhancers occurring at -4 and -10 kb relative to the transcriptional start site [89,90]. These enhancers contain sequences homologous to the antioxidant responsive element (ARE) [89,90]. The NF-E2-related factor-2 (Nrf2) represents the major transcriptional regulator of hmox-1 in response to many inducing stimuli [91,92]. Following cellular stimulation, Nrf2 migrates to the nucleus where it recognizes ARE binding sites in the hmox-1 promoter [93]. Nrf2 is inactivated by a cytoplasmic anchor, Keap-1 [93,94], and antagonized in the nucleus by a heme-sensitive transcriptional repressor, Bach-1 [95,96]. Additionally, a number of diverse transcriptional regulators can regulate hmox-1 in a cell-type and inducer-specific fashion [97,98]. These include hypoxia-inducible factor-1, heat shock factor-1 (HSF-1), activator protein-1, early growth response factor-1 (Egr-1), nuclear factor kappa-B (NF-κB), and others [7,84,97,98].
CO AS AN EFFECTOR OF INTRACELLULAR SIGNAL TRANSDUCTION PATHWAYS
Despite the known toxicity of CO at high concentration, recent research has revealed that low concentrations of CO may influence intracellular signal transduction pathways. CO can exert vasoregulatory properties [47], as well as modulate inflammation, apoptosis, and cell proliferation in vitro and in vivo [7,11,46].
Cellular exposure to CO has been shown to directly or indirectly modulate the activity of several intracellular signaling molecules (Fig. 2). Similar to NO, CO can act as a heme-ligand and activator of sGC, to increase the production of cGMP [99]. Experimental evidence indicates that NO activates sGC in vitro and corresponding vasodilatory action in vivo with greater potency [100].
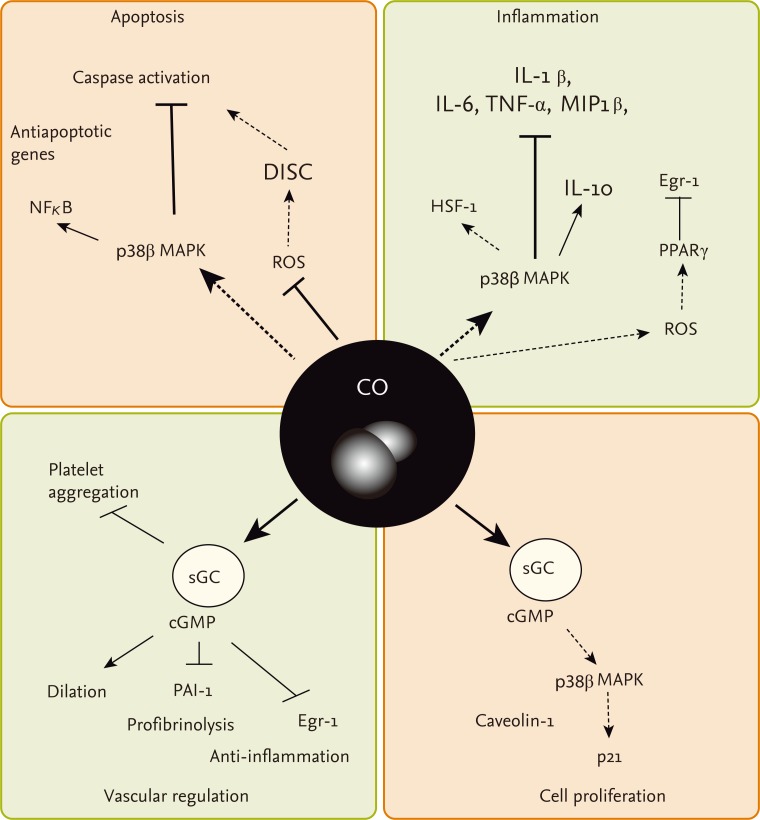
Signaling pathways regulated by carbon monoxide (CO). CO can confer modulatory effects on the regulation of vascular function, inflammation, apoptosis, and cell proliferation, through stimulation of several signaling pathways. The sGC/cGMP axis has been implicated in vascular effects of CO with respect to vessel dilation, regulation of platelet aggregation, and regulation of fibrinolysis through PAI-1. The sGC/cGMP axis has also been implicated in downregulation of cell proliferation by CO, through upregulation of p38 mitogen activated protein kinase (MAPK) and p21Waf1/CIP1. Anti-inflammatory and antiapoptotic effects of CO, including downregulation of proinflammatory cytokines production are also thought to be mediated by p38 MAPK. Additional mechanisms involving the inhibition of cytosolic reactive oxygen species (ROS) may play a role in regulation of apoptosis through inhibiting death-inducing signal complex (DISC) formation. Stimulation of mitochondrial ROS may upregulate PPARγ leading to downregulation of the proinflammatory factor Egr-1. Additional signaling molecules such as heat shock factor-1 (HSF-1) and caveolin-1 have been shown to mediate the anti-inflammatory and antiproliferative effects of CO, respectively. IL, interleukin; TNF, tumor necrosis factor; MIP1, macrophage inflammatory protein 1; NF-κB, nuclear factor kappa-B; Egr, early growth response; PPAR, peroxisome proliferator-activated receptor; sGC, soluble guanylate cyclase; cGMP, cyclic guanosine monophosphate.
CO was first implicated as a regulator of the sGC/cGMP axis in the context of olfactory neurotransmission [44]. CO can exert vasorelaxant effects of CO in the liver and other vascular beds which are believed to be dependent of cGMP [47,48,101]. Alternatively, CO may also regulate vascular function through additional proposed mechanisms including inhibition of cytochrome p450, and the activation of calcium-dependent potassium channels Kca in vascular smooth muscle cells [102].
CO can modulate the activation of the mitogen activated protein kinases (MAPK), which are important mediators of inflammatory and stress responses [43,45,53,55,56]. A potent anti-inflammatory effect of CO was demonstrated in bacterial lipopolysaccharide (LPS)-stimulated macrophages, that depended on the modulation of mitogen activated protein kinase kinase-3 (MKK3)/p38 MAPK pathway [43]. The anti-inflammatory effects of CO also may depend on the downregulation of Toll-like receptor trafficking and activation, the inhibition of NADPH: oxidase dependent signaling [103], and additional signal transduction pathway molecules including HSF-1, and Egr-1/PPARγ [104,105].
CO has been shown to modulate apoptotic signaling pathways in cultured cells [45,55,56]. When applied at low concentration, CO inhibited cell death caused by proapoptotic agents (e.g., tumor necrosis factor-α [TNF-α]) in endothelial cells, which required the p38 MAPK pathway, and modulation of NF-κB signalling [106]. Additional targets for CO-dependent regulation of apoptosis in cells subjected to oxygen-dependent stress include the STAT3 and phosphatidylinositol-3-kinase/Akt pathways [107], downregulation of NADPH: oxidase dependent reactive oxygen species formation [108], and modulation of Fas-initiated extrinsic apoptotic pathways [55,108,109].
CO also has been shown to exert antiproliferative effects in vitro, with respect to the proliferation of vascular smooth muscle cells (SMCs) [49,110,111]. In SMC, both sGC/cGMP and p38 MAPK signaling pathways have been implicated in the antiproliferative effects of CO these cells [49,58,110,111].
Additional signaling mechanisms potentially involved in CO-dependent regulation of cell proliferation, include the regulation of the lipid-raft associated signaling protein caveolin-1 [112], and modulation of NADPH oxidase [113]. More recent studies have implicated the NOX1 isoform of NADPH oxidase in CO dependent inhibition of SMC migration [114]. Taken together, these modulatory effects of CO on the signal transduction pathways that culminate in the regulation of inflammation, apoptosis, cell proliferation, and vascular function all may contribute to the proposed therapeutic effects of this gas.
CO AS AN INHALATION THERAPEUTIC: PRECLINICAL STUDIES
The therapeutic benefit of CO inhalation has been shown in a number of preclinical animal models of lung and vascular disease, as outlined in the following sections.
Endotoxin challenge
Anti-inflammatory effects of CO have been demonstrated in a mouse model of endotoxin exposure [43]. CO preconditioning reduced the production of serum TNF-α, interleukin (IL)-1β, IL-6, whereas increased the production of the anti-inflammatory cytokine IL-10; reduced organ injury and prolonged survival following LPS challenge [43,115]. Anti-inflammatory effects of CO with respect to modulation of pro or anti-inflammatory cytokines production were diminished in HSF-1 knockout (hsf1-/-) mice, implicating a role of the heat shock response in vivo [104].
Anti-inflammatory effects of CO have been recently documented in higher mammals including swine. CO reduced the development of disseminated intravascular coagulation and inhibited serum levels of the proinflammatory IL-1β in response to LPS, while inducing the anti-inflammatory cytokine IL-10 after LPS challenge [116]. Similar, though less significant effects were noted in a nonhuman primate model of Cynomolgous macaques subjected to LPS challenge [117]. CO exposure (500 ppm, 6 hours) following LPS inhalation decreased TNF-α release in broncoalveolar lavage fluid (BALF), but had no apparent effect on IL-6 and IL-8 release, in addition to reducing pulmonary neutrophilia (not observed at lower concentrations of CO). This reduction of pulmonary neutrophilia was comparable to pretreatment with a well characterized inhaled corticosteroid budesonide. However, the therapeutic efficacy of CO required relatively high doses that resulted in elevated COHb levels (>30%). This work highlights the complexity of interspecies variation in lung responses to CO, and of dose-response relationships of CO to COHb levels and anti-inflammatory effects [117]. This study was the first to examine the therapeutic index and dose-response relationships of CO therapy in nonhuman primates [117].
ALI
Low doses of CO have been shown to provide protection against ALI in rodent models. The administration of CO (250 ppm) during hyperoxia exposure prolonged the survival of rats and mice subjected to a lethal dose of hyperoxia, and dramatically reduced histological indices of lung injury, including airway neutrophil infiltration, fibrin deposition, alveolar proteinosis, pulmonary edema, and apoptosis, relative to animals exposed to hyperoxia without CO [52,53]. In mice, hyperoxia was shown to induce the expression of proinflammatory cytokines (i.e., TNF-α, IL-1β, IL-6) and activate major MAPK pathways in lung tissue. The protection afforded by CO treatment against the lethal effects of hyperoxia correlated with the inhibited release of proinflammatory cytokines in the BALF. The protective effects of CO in this model were found to depend on the MKK3/p38β MAPK pathway [53]. It should be noted that some studies have reported negative findings with respect to protective effects of CO in rodent ALI models [118,119]. More recent studies report protective effects of HO-1 or CO in a model of hyperoxia-induced bronchopulmonary dysplasia in neonatal mice [120]. Lung specific transgenic overexpression of HO-1 alleviated hyperoxia-induced lung inflammation, edema, arterial remodeling, and right ventricular hypertrophy. Similar protective responses were elicited by intermittent CO inhalation in this model. However, neither CO nor HO-1 expression prevented alveolar simplification in this model [120].
Ventilator-induced lung injury (VILI)
The therapeutic potential of CO has been shown in a specialized clinically-relevant model of VILI [54,121-123]. Rats ventilated with an injurious (high tidal volume) ventilator setting in combination with LPS injection, exhibited lung injury. The inclusion of CO (250 ppm) during mechanical ventilation reduced the inflammatory cell infiltration in BALF. In the absence of cardiovascular effects, CO dose-dependently decreased TNF-α and increased IL-10 in the BALF [54]. CO application has also been reported to confer tissue protection in a mouse model of VILI, at moderate tidal volume ventilation [121-123]. In this model, mechanical ventilation caused significant lung injury reflected by increases in protein concentration, total cell and neutrophil counts in BALF. CO reduced mechanical ventilation-induced cytokine and chemokine production and prevented lung injury during ventilation, involving the inhibition of mechanical ventilation-induced increases in BALF protein concentration and cell count, lung neutrophil recruitment, and pulmonary edema [121-123]. To date, these effects of CO were associated with the activation of caveolin-1 [121], activation of PPARγ, and the inhibition of Egr-1 signaling [122]. These studies, taken together, suggest that mechanical ventilation in the presence of CO may provide protection in animal models of VILI. However, more studies are required to determine the exact mechanisms underlying the therapeutic potential of CO in VILI models.
Organ ischemia reperfusion injury and transplantation
Tissue protective effects of CO have been shown experimentally in rodent models of organ I/R injury. Lung I/R caused by occlusion of the pulmonary artery causes lung apoptosis, involving caspase activation, expression changes in Bcl2 family proteins, cleavage of PARP, and mitochondrial cytochrome c release [55]. CO conferred tissue protection in rodents subjected to lung I/R injury, as evidenced by reduced markers of apoptosis, which depended on activation of the MKK3/p38α MAPK pathway [56]. In vivo studies using homozygous HO-1 knockout mice (hmox-1-/-) demonstrated that HO-1 deficiency conferred sensitivity to the lethal effects of lung I/R injury. Application of exogenous CO by inhalation compensated for the HO-1 deficiency in hmox-1-/- mice, and improved survival during pulmonary I/R [124]. The protection provided by CO involved activation of fibrinolysis. This effect of CO depended on activation of the sGC/cGMP axis, and downstream inhibition of plasminogen activator inhibitor-1, a macrophage-derived activator of SMC proliferation [124]. CO also inhibited fibrin deposition and improved circulation in ischemic lungs [125], involving the downregulation of the proinflammatory transcription factor Egr-1 [125]. Recent studies in a retinal I/R injury model, suggest that postinjury application of CO can inhibit injury of retinal ganglion cells through antiapoptotic and anti-inflammatory effects [126].
In organ transplantation models, I/R injury subsequent to transplantation, may play a major role in graft failure. In this regard, CO has been intensively studied as an anti-inflammatory therapeutic in experimental organ transplantation. CO has a demonstrated potential for reducing transplant associated I/R injury and also reducing the probability of graft rejection when applied at low concentration, as an adjuvant to organ preservation fluid or when applied to donors and/or recipients in gaseous form at low concentration. The application of CO has now been shown to confer protection during transplantation of several organs, including vascular tissue [58], heart [59], small intestine [127], kidney, [128], liver [129], and lung [60,130,131]. During orthotopic left lung transplantation in rats, exogenous application of CO (500 ppm), significantly protected the graft, and reduced hemorrhage, fibrosis, and thrombosis after transplantation [60]. Furthermore, CO inhibited lung cell apoptosis and downregulated lung and systemic proinflammatory cytokine production which were induced during transplantation [60]. Furthermore, protection against I/R injury was conferred in syngeneic rat orthotopic lung transplantation by inhaled CO administered to either the donor or the recipient [130]. Delivery of CO to lung grafts by saturation of the preservation media reduced I/R injury and inflammation in syngeneic rat orthotopic lung transplantation [131]. In a vascular transplantation model, when transplant recipients of aortic grafts were maintained in a CO environment (250 ppm) with preconditioning, these animals displayed reduced intimal hyperplasia, and reduced leukocyte, macrophage, and T cell infiltration in the graft [58]. Saturation of the organ buffer with CO gas also prevented cold I/R injury during subsequent intestinal transplantation [132]. The inhibition of apoptosis and inflammation may represent the primary mechanisms by which CO protects transplanted organs from dysfunction and failure [133], though improvement of blood circulation by CO within the reperfused transplanted organ [58,133,134], as well as antiproliferative [58] effects may contribute.
Pulmonary hypertension
Pulmonary arterial hypertension (PAH) is a terminal disease characterized by a progressive increase in pulmonary vascular resistance leading to right ventricular failure. Several previous studies have demonstrated that HO-1 expression can exert a protective effect in animal models of pulmonary hypertension, and in the regulation of hypoxic pulmonary vasoconstriction [135-137]. Administration of CO was shown to provide protection in rodent models of monocrotaline-induced and hypoxia-induced PAH. Exposure to CO (1 hr/day) reversed established PAH and right ventricular hypertrophy, restored right ventricular and pulmonary arterial pressures, as well as pulmonary vascular morphology, to that of controls. The ability of CO to reverse PAH was dependent on endothelial NOS3 and NO generation, since CO failed to reverse chronic hypoxia-induced PAH in mice genetically deficient for eNOS (nos3-/-). The protective effect of CO was endothelial cell-dependent, and associated with increased apoptosis and decreased cellular proliferation of vascular SMCs [138]. Additional studies have shown that CO decreased pulmonary artery vascular resistance and inhibited hypoxic vasoconstriction, through activation of the sGC/cGMP, and the hyperpolarization of potassium channels [139].
Fibrotic lung disease
Idiopathic pulmonary fibrosis (IPF) is a terminal disease characterized by scarring or thickening of lung tissues associated with fibroblast hyperproliferation and extracellular matrix remodeling with no known etiology or effective treatment [140]. IPF affects primarily the lower respiratory tract resulting in compromised efficiency of alveolar gas exchange [140]. Bleomycin, a redox cycling compound that generates O2- and H2O2, causes lesions in mouse lung after intratracheal administration, is used to model IPF in animals. Exogenous CO treatment can provide protection against bleomycin-induced fibrotic lung injury in mice [141]. In mice treated with bleomycin intratracheally and then exposed to CO or ambient air, the lungs from CO-treated animals displayed reduced lung hydroxyproline, collagen, and fibronectin levels relative to air-treated bleomycin-injured controls. The protective effect of CO in this model was associated with an anti-proliferative effect of CO on fibroblast proliferation associated with the increased expression of p21Waf1/Cip1 and inhibition of cyclins A/D expression [141].
Diabetes and metabolic syndrome
To date, only few studies have examined the direct therapeutic effects of CO in diabetes-related models. Diabetic gastroparesis is a condition where gastric emptying time is delayed, which is associated with increased oxidative stress, and injury to interstitial cells of Cajal in the stomach [142]. Nonobese diabetic mice with a gastric delay phenotype were subjected to inhaled CO therapy (100 ppm, 8 hr/day for 16 days), which reduced serum oxidative stress markers, restored expression of Kit, a marker of interstitial cells of Cajal, and ameliorated gastric delay in this model [143]. Similar effects were previously observed in this model with HO-1 overexpression [142]. Previous studies have suggested an antioxidative role for CO (administered as donor compound) in preventing hyperglycemia-induced endothelial cell sloughing in streptozotocin-induced diabetes [144].
In a type 1 diabetes models, ex vivo treatment of dendritic cells with gaseous CO was shown to augment dendritic-cell based therapy. Application of CO-conditioned dendritic cells was shown to effectively impair the accumulation and pathogenic activity of autoreactive CD8+ T cells in the pancreas [145].
CO exposure inhibited apoptosis in cultured pancreatic β-cells exposed to proapoptotic stimuli, through activation of the sGC/cGMP axis [146]. Similar to results reported in other transplantation models, CO preconditioning of mouse islets or treatment of donors improved viability and reduced graft rejection during allogeneic islet transplantation [146,147].
In models of metabolic disease, inhalation of CO gas reduced hepatic steatosis in mice subjected to 30% fructose or methionine-deficient and choline-deficient diets [148]. CO exposure (administered as donor compound) was shown to confer cardioprotection and restore mitochondrial function in a high fat diet induced model of metabolic syndrome [149]. Taken together, these studies are suggestive of a potential for CO therapy of metabolic disorders, though further investigation is needed.
Preeclampsia
Preeclampsia is a condition associated with pregnancy involving abnormal placentation, hypertension, and proteinuria. Although the condition is thought to involve increased oxidative stress in the placental circulation, women who smoke during pregnancy have a significantly reduced risk of developing this condition [150]. Previous studies have suggested that deficiency in the HO-1/CO system may be associated with placental dysfunction and susceptibility to preeclampsia [151]. HO-1 expression was found to be reduced in placenta from pregnancies complicated by preeclampsia [151]. Consistently, blood COHb levels were found to be significantly lower in women with preeclampsia compared with normal pregnancies [152]. Ex vivo application of CO to placental villous extracts reduced I/R associated apoptosis in the syncytiotrophoblast layer [153]. Intriguingly, recent clinical studies have described ambient CO as an inverse risk factor for preeclampsia. Maternal exposure to moderate ambient CO was associated independently with a decreased risk of preeclampsia [154]. Current views implicate the HO-1/CO system as an essential procirculatory factor in the placenta, though more studies are needed.
PHARMACOLOGICAL APPLICATION OF CO USING DONOR COMPOUNDS
As an alternative approach to the administration of CO gas by inhalation, chemical CO-donor compounds termed carbon monoxide releasing compounds (CORMs) have been developed as experimental therapeutics over the last decade [11].
Several prototypical CORMs have been extensively tested in experimental models, including the original Mn2CO10 (CORM-1) and the ruthenium compounds tricarbonyldichlororuthenium-(II)-dimer (CORM-2) and tricarbonylchoro (glycinato)-ruthenium (II) (CORM-3) [155,156].
CORM-1 and CORM-2 are hydrophobic, while CORM-3 is water-soluble and rapidly releases CO in physiological fluids. A water-soluble boron-containing CORM (CORM-A1) has also been developed, which slowly releases CO in a pH and temperature-dependent fashion [157]. A new CORM (CORM-S1) based on iron and cysteamine has recently been synthesized, which is soluble in water and releases CO under irradiation with visible light, while it is stable in the dark [158]. Micellar forms of metal carbonyl complexes have been developed that display slower kinetics of CO release and enhanced ability to target distal tissue drainage sites [159]. Furthermore, novel hydrophobic and hydrophilic CORMs based on iron carbonyls have been recently described [160].
CORMs have demonstrated vasoactive effects with CORM-3 shown to produce a rapid vasodilatory response [161]. Similar to inhalation CO, cytoprotective effects have been obtained in various injury and disease models with pharmacological application of CORMs. CORMs can be used to deliver small amounts of CO to biological systems in a controlled manner and are emerging as an experimental therapy for sepsis and inflammatory disorders. An advantage of CORMs is that they deliver CO to tissues with less COHb buildup typical of inhalation CO [11].
CORMs have been shown to inhibit proinflammatory cytokine production in LPS-stimulated macrophages [162], and decrease the inflammatory response and oxidative stress in LPS-stimulated endothelial cells [163]. In vivo, CORMs attenuate systemic inflammation and proadhesive vascular cell properties in septic and thermally injured mice by reducing nuclear factor-κB activation, protein expression of ICAM-1, and tissue granulocyte infiltration [164,165]. CORM-3 has been shown to prevent reoccurrence of sepsis, CORM-2 prolongs survival and reduces inflammation and CORM-3 reduces liver injury after CLP [165,166]. Recent studies on the protective effects of CORMs in murine sepsis were related to stimulation of mitochondrial biogenesis through the Nrf2/Akt axis [167]. Furthermore, in cardiac transplantation model, inclusion of CORM-3 in the preservation fluid improved cardiac function following transplantation [168].
These studies taken together have demonstrated that the CORM dependent release of CO can confer protection in models of inflammation and sepsis, suggesting that CORMs could be used therapeutically to prevent organ dysfunction and death in sepsis.
CO AS AN INHALATION THERAPEUTIC: CLINICAL STUDIES
To date, inhalation CO has been administered to humans in but a few published experimental studies. CO inhalation was administered to healthy human subjects to examine systemic inflammation during experimental endotoxemia. In a randomized, double-blinded, placebo-controlled, two-way cross-over trial, experimental endotoxemia was induced in healthy volunteers by injection of 2 ng/kg LPS. The potential anti-inflammatory effects of CO inhalation were investigated by inhalation of 500 ppm CO (associated with an increase in COHb from 1.2% to 7%) versus synthetic air as a placebo for 1 hour. In this study, CO inhalation had no effect on the inflammatory response as measured by systemic cytokine production (TNF-α, IL-6, IL-8, IL-1α, and IL-1β), although no adverse side effects of CO inhalation were observed [169]. However, given the limited scope of this initial trial, and the protective characteristics of CO application in many animal models of sepsis, further more detailed clinical trials are required to reach a verdict on the efficacy of CO for reducing inflammation in septic patients. In contrast, a recent clinical study demonstrates the feasibility of administering inhaled CO to humans with COPD [170]. In this study, exsmoking patients with stable COPD were subjected to CO inhalation (100 to 125 ppm for 2 hr/day for 4 days), which increased COHb levels to 4.5%. Inhalation of CO by patients with stable COPD led to trends in reduction of sputum eosinophils and improvement of methacholine responsiveness [170].
CONCLUSIONS
Gaseous molecules continue to show future promise as they join the armamentarium of experimental and clinical therapeutics. Among the known medical gases, CO inhalation has been demonstrated to have potential applications in pulmonary diseases and other inflammatory diseases. To date, salutary effects of CO have been demonstrated in a number rodent model studies, though recent studies have attempted to recapitulate findings in larger animals such as monkeys and swine [5,7,116,117]. Differences in lung physiological responses to CO between rodents, large animals (e.g., nonhuman primates) and humans require further investigation. Additional studies will be required to confirm the safety and efficacy of CO inhalation as a treatment for inflammatory lung diseases. Pharmacological application of CO using CORM technology provides an attractive alternative to inhalation gas [11]. However, further understanding of the pharmacokinetics and toxicological responses of CORMs, including hemodynamic effects, must be achieved before employing CORMs as clinical therapy. The effectiveness of inhaled CO as a therapeutic in human diseases including sepsis, renal transplantation, pulmonary fibrosis, and pulmonary hypertension, awaits the outcome of additional planned preclinical testing and clinical trials. The next decade should yield a resolution on the feasibility and efficacy of exploiting the therapeutic index of CO in human disease.
Acknowledgments
This work was funded by NIH grants P01 HL108801 and R01 HL079904 to A.M.K. Choi. S.W. Ryter received additional salary support from the Brigham and Women's Hospital and Lovelace Respiratory Research Institute Consortium for Lung Research.
Notes
No potential conflict of interest relevant to this article is reported.