Roles of mast cells in rheumatoid arthritis
Article information

Abstract
Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory arthritis, and the complex interaction and activation of innate and adaptive immune cells are involved in RA pathogenesis. Mast cells (MCs) are one of the tissue-resident innate immune cells, and they contribute to RA pathogenesis. In the present review, the evidence of the pathologic role of MC in RA is discussed based on human and animal data. In addition, the potential role of MC in RA pathogenesis and the research area that should be focused on in the future are suggested.
INTRODUCTION
Rheumatoid arthritis (RA) is a systemic autoimmune arthritis that affects small peripheral joints. Inflammatory changes of synovium (synovitis) is a cornerstone characteristic of RA, and synovial hyperplasia with the infiltration of synovial membrane with various immune cells, T cells, B cells, and innate immune cells such as mast cells (MCs), have been observed in RA synovitis [1]. In RA, uncontrolled synovitis leads to the progressive destruction of joint structures including cartilage and bone, and thus, prevention of structural damage in the joints is one of the most important treatment targets. Many synthetic disease-modifying antirheumatic drugs (DMARDs) have therapeutic effects, and nowadays, several biologic DMARDs targeting monocytes, T cells, and B cells and oral small molecule inhibitors such as Janus kinase inhibitors are used in managing RA patients who do not respond to synthetic DMARDs. Despite novel therapeutic options, biologic DMARDs, and oral small molecule inhibitors, some patients still do not respond to these therapies. Furthermore, the prevention of RA in preclinical or early stages of RA has been attempted, and research on this theme has been ongoing [2,3]. However, administering synthetic or biologic DMARDs in the early phase of RA does not have dramatic effects on preventing RA.
Post-translational modifications such as citrullination and carbamylation induce aberrant immune response via antigen presenting cells (APCs) and promote autoantibody production [3]. The RA patients that have autoantibodies, immunoglobulin M (IgM) to Fc portion of IgG (rheumatoid factor), anti-citrullinated protein antibody (ACPA), or anti-carbamylated protein antibody, are called seropositive RA patients, and these autoantibodies play a critical role on RA pathogenesis [4]. Thus, RA was believed to be an autoimmune disease caused by aberrant adaptive immune response initiated by autoantibody production [5]. However, the pathologic role of MCs in RA emerged in a study that showed the histologic finding of synovium, which indicated an increased proportion of MCs in RA patients than those in healthy control [6]. Furthermore, the MCs interact with various immune cells and show the potential pathologic role of MCs in various autoimmune disorders [7]. Recently, the analysis of early stage RA synovium showed different phenotypes of synovitis according to the density of MC infiltration, thereby supporting that the underlying mechanisms of RA pathogenesis are heterogenic and MCs may play an important role in RA pathogenesis [8].
RA is divided into several stages: autoantibodies production, subclinical synovitis, and clinical established RA [9]. Immune response is initiated by local innate immune cells that are exposed to external antigens or autoantigens (such as MCs, macrophages, dendritic cells, and natural killer [NK] cells) in the case of autoimmune diseases [10]. The APCs then interact and activate the immune cells of the adaptive immune system such as B cells and CD4+/CD8+ T cells. Thus, the initial immune responses are dominant in local innate immune cells. MCs are one of the innate immune cells that are site specific and intimate with the external environment [11]. The MCs respond to the antigen before the other immune cells, and they secrete various chemokines, recruit other immune cells, and cause local tissue inflammation, edema, neovascularization, and tissue remodeling [11]. Furthermore, RA synovium shows increased vascularity and MC population than in a healthy synovium, which indicates the potential increase in the interaction between MCs and autoantigens in the early phase of autoimmune diseases. Based on previous results that showed an increased MC population in RA synovium, research investigating the role of MC in RA pathogenesis is ongoing. This review focuses on the pathologic roles of MC in RA and potential therapeutic alternatives for targeting MCs.
MAST CELLS IN RHEUMATOID ARTHRITIS PATHOGENESIS
Overall characteristics of MC
MCs were first found by Friedrich von Recklinghausen in 1863 and well defined by Paul Ehrlich in 1878. An MC is a tissue-resident cell originated from a myeloid stem cell [11]. The MC progenitors are derived from bone marrow, and they circulate in the blood stream. Settled MC progenitors completely differentiate into MCs in the peripheral tissues, and they typically contain preformed mediators such as histamine, proteases, and heparin [12]. Further, MCs have the high-affinity IgE receptor (FcεRI), and they act as the major immune cells in allergic reactions causing an immediate release of the preformed mediators [13]. Allergic and autoimmune diseases have similar characteristics in that they are results of aberrant and hyperactive immune response to allergens or autoantigens, which are not immunogenic in a normal immune system. Furthermore, many evidences indicate the pathologic role of MCs in autoimmune diseases including RA [7], and previous studies have shown an increase in the population of MCs in RA synovium [1,6].
MC in RA synovium
MCs exist in normal synovium; their population increases in RA synovium compared to that in healthy control [6]. The MC count correlates with clinical synovitis, but not with erythrocyte sedimentation rate (ESR) [6]. In another study, active RA shows higher MC infiltration than end-stage diseases [14]. The MC count shows strong correlation with lymphocyte count in RA synovium, and intra-articular steroid injection reduces the MC count in the RA synovium [15]. The morphology and ability of the histamine secretion of synovial MCs are similar to that of the MC from the lung and intestine, which indicates that synovial MC is not significantly different from the MC of other organs [16]. The MCs from RA synovium differ from those of the osteoarthritis (OA) synovium in that the C5a receptor (CD88) expression is solely present in RA synovial MC [17]. Stem cell factor (SCF), a ligand of c-kit, and c-kit positive MC hyperplasia are correlated with local inflammation in the synovium; however, this response occurs in not only RA, but also in traumatic arthritis or OA [18]. The RA synovial fluid shows elevated chemotaxin for the MC, SCF, and transforming growth factor-β (TGF-β) [19]. The MCs are divided based on their phenotypes according to expressing proteases, tryptase, and chymase. MCT only presents tryptase, whereas MCTC presents both tryptase and chymase. There is a higher infiltration rate of MCs in fibrous synovial tissues (2.8%) than in cellular synovial tissues (1.18%), and extracellular tryptase (which indicate the activation of MC) is associated with localized tissue edema and stromal matrix disruption [20]. In this study, MCT is more prominent than MCTC in RA synovium (MCT:MCTC ratio, 8:1) [20]. MCTC and MCT are all expanded in the superficial and deeper layers of RA synovium than that in the healthy synovium. MCT is dominant in the superficial layer, which is the area close to the inflammatory cell infiltration, whereas MCTC is located in a deeper layer, which is the area approximately close to the cellular, dense, fibrous connective tissues [21]. Furthermore, the total MCTC correlates significantly with the swollen joint counts, and the superficial MCTC correlates with the disease severity index [21]. In early RA (mean duration 8 months), MCT are dominant and they significantly correlate with the inflammation index [22]. These results suggest that MCT and MCTC might have different roles, such as MCT for the early inflammation response and MCTC for the late destructive change of RA [21,22]. All previous studies have some limitations in that most synovia are obtained from knee joints, whereas RA affects small joints, and the disease duration is relatively long.
Recently, utilizing ultrasonography (US) for the evaluation of arthritis, some studies based on US-guided synovial biopsy have been introduced. Synovial tissues from reactivated RA patients show increased MCs and B cells than those in RA patients who maintain remission, which indicates the potential role of MCs in disease reactivation [23]. In treatment-naïve RA patients, early RA can be divided into three subgroups: fibroid (low MC density), myeloid (medium MC density), and lymphoid (high MC density) [8]. MC density positively correlates with acute reactants (C-reactive protein, ESR) and disease activity score (DAS-28) [8]. The ratio of MCT and MCTC is different based on the subtypes (MCT:MCTC ratio in fibroid 1:6, lymphoid 1:2), and MCT is significantly correlated with synovial inflammation, indicating the potential role of MCT in the acute inflammation of RA [8,22].
ACTIVATION OF MAST CELL IN RHEUMATOID ARTHRITIS
MC is activated via various pathways, and the activated MC secretes mediators, stimulates other immune cells and local synoviocytes, and recruits circulating inflammatory cells into the RA synovium. Preformed mediators (histamine, heparin, and proteases) are excreted from MCs by immediate immune response; however, MCs are also capable of producing and excreting various lipid-derived mediators, cytokines, and chemokines [24]. The aforementioned de novo production is mediated by several immunologic stimulations.
MCs are activated by various stimulants such as the immunoglobulins-Fc receptor interaction, toll-like receptors (TLRs), chemokines, cytokines, pathogen-associated molecular patterns (PAMP), or neuropeptides [11]. The MCs are mainly activated via IgE stimulations, and MCs from RA are also stimulated by the IgE-FcεR interaction [16,17]. The MCs possess not only FcεR, but also Fc gamma receptor (FcγR), and therefore, they can be activated even with IgG, which is the major immunoglobulin produced under chronic inflammatory status such as RA.
Immunoglobulin-FcR interaction
Conventionally, MC activation is mediated via IgEFcεR interaction in in vitro experiments. Histamine, tryptase, proinflammatory cytokines (IL-1, tumor necrosis factor-α [TNF-α]), and lipid-derived mediators (prostaglandin D2 [PGD2]/prostaglandin E2 [PGE2] and leukotriene C4) secreted from RA synovial MC are augmented via anti-IgE stimulation [16,17,24-26]. Human synovial MC presents FcεRI, Fc-gamma receptor type 1 (FcγRI), and Fc-gamma type 2 receptor (FcγRII), but not Fc-gamma type 3 receptor (FcγRIII) [26], and the RA synovial MCs specifically overexpresses Fc-gamma type 2 receptor A (FcγRIIA) [27]. In human synovial MCs, FcγRI-IgG mediates PGD2 and TNF-α production, whereas histamine production is mediated via both FcγRI and FcγRII [26]. Furthermore, ACPA-citrulline peptide immune complex activates MC and promotes IL-8 secretion via FcγRIIA [27,28].
TLR ligands
TLRs are expressed in various immune cells, and they play a critical role in eliminating exogenous pathogens by recognizing PAMP. Endogenous TLR ligands, such as heat shock protein 70 (HSP70), are increased in the RA synovial fluid, and the TLR4 ligands, tenascin-C, and extra domain A of fibronectin, induce joint inflammation in a mice model [29-31]. TLR1-TLR9 are expressed in MC, and TLR2, TLR4, and TLR8 are capable of inducing TNF-α production [27]. MC activation via HSP70-TLR4 interaction is synergized when ACPA immune complex-FcγRIIA co-exists [27].
MC activation via neuropeptides and cytokines
Substance P—one of the neuropeptides—induces MC activation via Mas-related X2 expression in RA synovium [32]. Stimulation with IL-33—an IL-1 superfamily— is critical in the induction of collagen-induced arthritis (CIA) and autoantibody-induced arthritis (AIA) animal models via MC activation [33,34]. In human RA synovium, IL-33 and the ACPA immune complex also induce MC activation, and the combination of IL-33 and immune complex increases inflammation via the upregulation of IL-8 and TNF-α production [28,35]. Simultaneously, this mediates IL-10/histamine secretion and induces an immune modulatory effect by suppressing monocyte activation [28]. Furthermore, IL-33 stimulated MC overexpresses FcγRIIA, which can augment ACPA mediated MC activation [28].
EFFECTOR ROLES OF ACTIVATED MAST CELL
Roles of preformed mediators of MCs in RA
Histamine is a major preformed mediator in MCs, and it increases vascular permeability and recruits neutrophils in RA animal models [36]. FcRγ-expressing cells mediate histamine-induced vascular leaks, although the exact cell type of FcRγ-expressing cells is unknown; the candidate cell is an MC [36]. In RA synovium, histamine is released by the stimulation of anti-IgE, SCF, and C5a [16,17]. MCs extracted from an active RA patient’s synovium secrete greater histamine than that from an inactive RA patient’s synovium under stimulation with anti-IgE; histamine content decreases in MCs in DMARD-treated RA patients [37]. Histamine binds with four receptors (histamine receptors 1–4 [H1R–H4R4]), and among them, H4R plays a crucial role in arthritis development in K/BxN RA mice model [38]. In terms of osteoclasts, the main effector cells of the bone erosion of RA, histamine and H4R, promote receptor activator of nuclear factor-κB ligand (RANKL) expression and osteoclastogenesis [39]. In a fracture and osteoporosis mice model, the granular mediators of MCs, especially histamine, are critical for osteoclastogenesis [40].
MCT and MCTC are increased in RA synovium. Tryptase excreted from RA synovial MC enhances collagen degradation by activating latent synovial collagenase [41]. The mouse model of RA shows an increased level of MC restricted mast cell protease 6 (MCP-6) (corresponding to human tryptase β)/heparin complexes in synovium, and these complex induces inflammatory arthritis and even bone loss caused by an increase in the neutrophil chemotactic factors with the loss of cartilage-derived aggrecan [42,43]. Tryptase inhibition in the mouse model reduces late phase joint edema, chemokine (C-X-C motif) ligand 1 (CXCL1), and IL-6 production; however, the overall joint destruction is not attenuated [44]. Another protease, chymase, is also elevated in RA synovial fluid than OA, and it proposes to mediate synovitis by producing the active form of chemerin (chem 156F), a chemoattractant for NK cells, macrophages, and dendritic cells [45]. In the RA rat model, synovial chymase from RA rat was found to promote the proliferation of synovial fibroblasts and the expression of matrix metalloproteinase 9 (MMP-9), focal adhesion kinase (FAK), and p21, which are markers for cell migration and adhesion [46]. Chymase induces “tumor-like” characters of synovial fibroblasts in RA [46]. Chymase and tryptase are well known angiogenetic factors in tumorous conditions [47]. In addition to protease, MC secretes various proangiogenetic factors, vascular endothelial growth factors, basic fibroblast growth factors, TGF-β, TNF-α, and IL-8 [48]. Synovial fibroblasts of RA have “tumor-like” characteristics— invasion, destructing adjacent tissues, and increasing vascularity by forming pannus—and therefore, suppressing the “tumor-like” features of RA synovium by modulating MCs containing proteases, chymase, and tryptase, is considered a potential therapeutic target.
Lipid derivatives and proinflammatory cytokine from MC in RA
Lipid derivatives from arachidonic acid are increased in RA synovium and play an inflammatory role; drug targeting enzymes of arachidonic acid metabolism— cyclooxygenase inhibitors—are useful for controlling the pain caused in arthritis [49]. Proinflammatory cytokines, IL-1β, IL-6, IL-17, and TNF-α, are elevated in RA patients systemically and at inflamed synovium [50]. Several biologic DMARDs have therapeutic effects induced by blocking these proinflammatory cytokines [4].
The metabolites of arachidonic acid depend on the enzymes. Lipoxygenase mediates the production of leukotrienes, and cyclooxygenases mediates prostaglandins and thromboxane production [49]. Leukotriene B4 (LTB4), PGE2, and prostaglandin I2 (PGI2) induce an inflammatory response in RA pathogenesis [49]. The genetic deficiency for the receptor of LTB4, leukotriene B4 receptor 1 (BLT1), and BLT2, prevents arthritis in mouse models [51]. Although MC is the major source of LTB4 in IgE-mediated allergic response [52], the role of MC-derived LTB4 in RA inflammatory response is not elucidated. Under stimulation with rabbit IgG anti-human IgE on RA synovium, PGE2 is overexpressed [25]. PGD2 is released from RA synovium-derived MC through FcγRI activation [26]. The urinary metabolite of PGD2, tetranor prostaglandin D metabolite (T-PGDM), in RA patients are higher than those in the control, and it is suggested as a biomarker of MC activation in RA patients [53].
Several proinflammatory cytokines, IL-1, IL-6, IL17, and TNF-α, play critical roles in RA pathogenesis, and the targeted therapies for these cytokines show dramatic therapeutic effects on RA [4]. The secretion of TNF-α and IL-1β from RA synovial MC significantly is increased by stimulation with rabbit IgG antibodies for human IgE [54]. There is increased TNF-α production of cultured synovium-derived MCs via an IgG receptor, FcγRI [26]. In AIA model, MCs are critical for the initiation of arthritis via the production of IL-1, and MCs are activated via FcγRIII [55]. The major source of IL-17A in RA synovium is MCs, and not CD4+ T cell [56]. However, another study shows that there is no difference in the IL-17A positive synovial MCs in RA and OA [57]. In the K/BxN serum transfer RA model, c-Jun N-terminal kinase (JNK) 1 controls MC activation and FcγR-triggered IL-1β production of MCs [58]. The early depletion of MCs in the preclinical phase of the CIA model shows decreased serum levels of IL-6 and IL-17 and reduced arthritis incidence [59]. The transplant of a wild-type MC restores the induction of clinical arthritis and the production of proinflammatory cytokines (IL-17, IFN-γ, TNF-α) in IL-33 receptor deficient mice [33,34]. IL-8 and TNF-α secretion is enhanced by IL-33 stimulation in synovial MCs [35]. MC stimulation with the rabbit IgG antibody for human IgE increases TNF-α production in the RA synovial tissue, and this is suppressed by the c-kit tyrosine kinase inhibitor, imatinib [60]. Another study of the CIA mice model shows an increased number of TNF-α positive MC in inflamed synovium, and anti-allergic drug, tranilast, which effectively suppresses the TNF-α positive MC [61]. IL-8, a neutrophil chemotactic factor, is synergistically increased in RA synovial MC by stimulation with the combination of TLR and FcγRIIA mediated activation [27].
CELLULAR INTERACTION OF MAST CELL WITH OTHER INFLAMMATORY CELLS AND LOCAL SYNOVIOCYTES
Cell interactions with MCs in RA
MCs are involved in RA pathogenesis via direct roles such as secreting preformed mediators and newly synthesized inflammatory mediators; however, MCs also influence other immune cells or local synoviocytes. In RA synovium, MCs closely contact CD3+ T cells, CD14+ monocytes, and CD20+ B cells [28]. These indirect influences of MCs on localized or circulating cells are another important pathologic role in RA [62].
MC-lymphocytes interactions
B and T cells are major immune cells of the adaptive immune system, and they play crucial roles in RA pathogenesis. Interactions between MC and these lymphocytes increase the adaptive immune response in RA pathogenesis. The in vitro experiment of mice shows that MCs induce naïve and B cell receptors that stimulate B cell proliferation and activation by cell-to-cell contact; further, it promotes immunoglobulin secretion [63]. Synovial MC from treatment naïve RA patients enhances B cell activation, differentiation, and ACPA production [8]. The aforementioned interaction of MC and B cells occur in ectopic lymphoid tissue of synovium in a contact-mediated manner [8]. These findings are worthy in that MC plays a critical role in autoantibody production in RA patients.
T cells are usually stimulated by APCs, such as dendritic cells, macrophage, and B cells. The T cell receptor (TCR) recognizes the antigen presented in APCs, and it becomes active. Stimulated MC expresses MHC molecules and acts as APC to CD4+ and CD8+ T cells [64-66]. MC can skew CD4+ T cell into a specific subtype of CD4+ T cell in a cell-to-cell contact manner [65,67]. The MCs can recruit effector CD8+ T cell via excreting LTB4 [68], and it also activates T cells by TNF-a production [69]. However, direct evidences supporting the importance of MC-T cell interaction in RA pathogenesis is yet to be proved.
Synoviocyte stimulation via MC activation
Cartilage and bone erosion with pannus formation are classical changes in advanced RA, and this is mediated by synovial fibroblasts. Tryptase from MC induces collagen degradation by transforming RA synoviocyte procollagenase into the active form via MMP-3 activation [41,70]. In addition, activated MCs are localized to the sites of cartilage erosion and matrix disruption in RA synovial species indicating that the activation of MCs is involved in the process of erosion [20,71]. Tryptase-β/heparin complex, one of the MC mediators, induces IL-8 expression in synoviocyte and promotes neutrophil recruitment [42]. Tryptase from MCs exert an anti-apoptotic effect on RA fibroblast-like synoviocytes (FLS) via rho-mediated signaling [72]. In the RA rat model, chymase from MC induces FLS proliferation and MMP-9/IL-6 expression, and this response is amplified in RA synoviocyte than that in normal synoviocyte [46]. This suggests that RA synoviocytes may have an augmented response to MC mediators compared to normal synoviocytes.
IMMUNOMODULATORY EFFECTS OF MAST CELL IN RHEUMATOID ARTHRITIS
The MC is a tunable cell and can also provoke anti-inflammatory immune response under certain circumstances [73]. In RA synovium, IL-33 induces MC activation and suppress monocytes via IL-10 and histamine [28]. MC specific gene expressions in RA synovium demonstrate inverse correlation with disease activity, and IL-33 mRNA levels inversely correlate with the mRNA levels of proinflammatory markers [28]. Serum and synovial mRNA levels of tryptase also inversely correlate with C-reactive protein levels in treatment naïve early RA patients [74,75]. The aforementioned results support that MCs may play both proinflammatory and anti-inflammatory roles in RA; however, the specific roles of MC mediators and MC responses in the specific stage of RA should be evaluated.
ROLE OF MAST CELL IN THE INDUCTION OF ARTHRITIS IN RHEUMATOID ARTHRITIS ANIMAL MODELS: CONTRADICTORY RESULTS
Evidences supporting pathologic roles of MC in RA: pros
Most animal models used to reveal the pathologic role of MC in RA are provoked via a conditional knockout of specific genes. Knockout mice of MC related to protease chymase, and tryptase/heparin complex genes demonstrate the suppression of arthritis in MC protease deficient mice [42,43,76]. CIA induction in A20 deficient mice, MC hyperactivity via ablating nuclear factor-κB negative feedback regulator, arthritis incidence and severity are more severe than that in the wild type control, which supports the pathologic role of MCs in RA [77]. Another approach is to eliminate MCs from mice by modulating SCF and c-kit related genes. SCF deficient (KitlSl/KitlSl-d) or SCF receptor (c-kit) deficient mice (KitW/KitW-v) have resistance to arthritis in K/BxN serum transfer model [78]. This KitW/KitW-v mice restore susceptibility to develop arthritis via intraperitoneal or intra-articular injection of MCs [79]. Mutation on c-kit signaling related genes can cause several immune and non-immune abnormalities beyond MC deficiency because it is not uniquely expressed in MCs. Diphteria toxin (DT) induced MC depletion is an alternative mice model for MC deficiency [80]. Mcpt5-Cre iDTR mice, which induces MC deficiency without altering c-kit signaling, reveals that MC deficiency reduces arthritis in antigen-induced arthritis mice (T cell dependent model), whereas MC is redundant in the pathogenesis of antibody-induced arthritis mice (T cell independent model) [81]. By using other DT induced MC depletion model, depleting MCs in established arthritis do not influence on arthritis progression, whereas early depletion of MC reduces clinical arthritis score in CIA model [59]. These findings support that MC may possess different importance according to disease stages, essentially in the early stage (prior to adaptive immune system activation and auto-antibodies production), but it is dispensable in the late stage of RA pathogenesis.
Redundant role of MC in RA pathogenesis: cons
Another c-kit mutation induced MC deficiency model, KitW-sh/KitW-sh mice, is susceptible for arthritis both in antibody mediated and antigen mediated models [82,83]. KitW/KitW-v mice and KitW-sh/KitW-sh mice have differences in that KitW/KitW-v mice have more clinical manifestations other than MC deficiency [84]. Importantly, KitW/KitW-v mice show neutropenia and attenuated response to lipopolysaccharide stimulation, whereas KitW-sh/KitW-sh mice have neutrophilia [82]. The baseline neutrophilia of KitW-sh/KitW-sh mice may contribute to the susceptibility of arthritis induction, and this makes MC dispensable in the KitW-sh/KitW-sh mice arthritis model. In Cpa3Cre/+ mice, MC depletion is achieved by Cre-recombinase, and arthritis can be induced by K/BxN serum transfer [85]. Cpa3Cre/+ mice have a normal immune system except MC deficiency, and this selective MC deficiency is different from that in c-kit signal mutant mice. These contradictory results of MC roles in animal models should be interpreted carefully by considering background mutation combined with other immune abnormalities. The roles of MC in RA pathogenesis proved in human and animal RA data are summarized in Table 1.
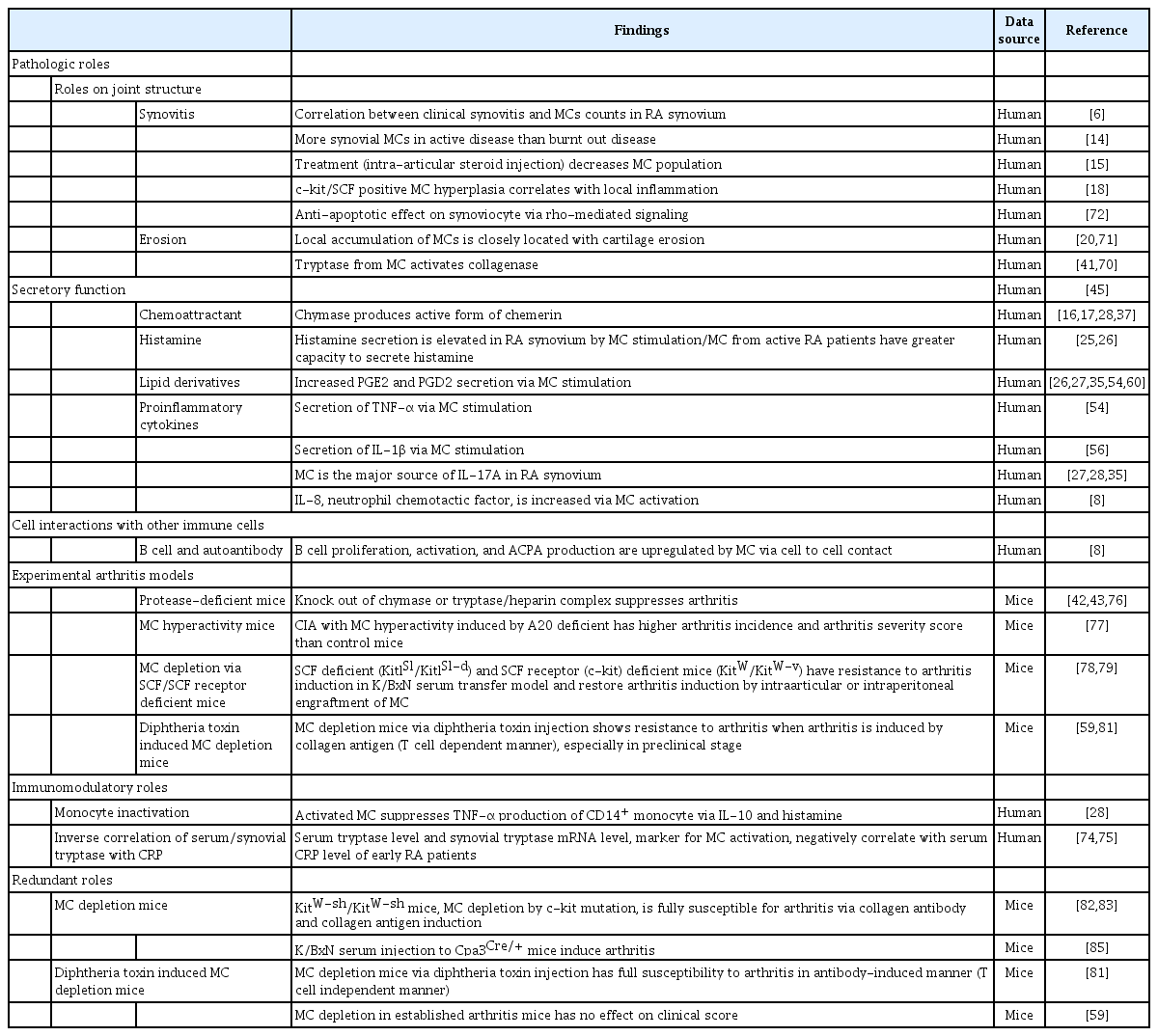
Evidences from human and animal RA data: roles of MCs in RA pathogenesis
CLINICAL IMPLICATION OF MAST CELL IN RHEUMATOID ARTHRITIS
Early RA is divided into three histological types according to synovial MC counts: fibroid, myeloid, and lymphoid types [8]. RA is heterogeneous disease, and each RA patient has different clinical manifestation, drug response, and disease course. Furthermore, applying “precision medicine” to RA patients has emerged [86], and the personalized treatment strategy aims to achieve early remission and cease structural damage of RA. Categorization of synovial pathology according to MC population suggests potential to establish “precision medicine” to RA.
In pharmacologic intervention research, imatinib, which is used in Philadelphia chromosome positive leukemia and inhibits c-kit tyrosine kinase, induces MC apoptosis and suppresses TNF-α production [60]. In animal model, applying MC stabilizer, cromolyn, salbutamol, and tranilast, suppress proinflammatory cytokine production and structural damages [61,79]. When comprehensively consider these experimental intervention and histologic type of RA synovium according to MC population, MC suppressor or stabilizer could promise adjuvant therapeutic effects for RA patients with MC rich in synovium (lymphoid type).
FUTURE RESEARCH AGENDA
Although previous studies demonstrated many evidences that showed pathologic roles of MC in RA pathogenesis, there were still many unrevealed roles of MCs. First, MCs secrete chemokines and derive infiltration of various immune cells such as neutrophils, T cells, and macrophages [87]; however, the direct evidence of these functions in RA is yet to be fully demonstrated. Further studies focusing on the chemoattractive function of MCs need to be performed. Second, MCs stimulate B cell responses in RA synovium [8], but MCs also stimulate various other immune cells, such as T cells, via acting as APCs [64,66,88]. However, the relevant roles of MCs in stimulating other immune cells are poorly understood in RA, except for B cells. Third, the influence of MCs in the angiogenesis of RA synovium needs to be studied. Several mediators, such as tryptase and chymase, promote neovascularization in various cancers [47], and these might have a role on forming “tumor-like” aggressive characteristics of RA synovium. Finally, ablating or suppressing MCs at the preclinical or early phase of RA might be an effective treatment choice in the future because several evidences support that MCs are critical in early RA pathogenesis. Several trials are performed to suppress MCs in RA synovium or RA mice [60,61,79], and these showed the potential of the therapeutic role of MC suppression in RA treatment. Table 2 summarizes the potential roles of MCs in RA pathogenesis and future research objectives that should be proved in RA.

Potential role of MCs in RA pathogenesis and future research agenda
CONCLUSIONS
The network of innate and adaptive immune systems plays critical roles in RA pathogenesis. The MCs belong to innate immune cells, but they also influence the adaptive immune system in various ways, such as by cell-to-cell contact or secreting cytokines and chemokines. Many evidences support the pathologic roles of MC in RA, especially recent US-guided synovial biopsy studies, which showed the correlation between disease activity and MC infiltration [8,23]. These studies suggest that MC might play a critical role in the onset of RA via interaction with B cells. Therefore, focusing on the role of MCs and modifying MCs in early or preclinical phases of RA might provide new insight into RA pathogenesis and novel treatment strategies.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This research was supported by a grant of the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology, Republic of Korea (NRF-2018R1D1A1A02050982).