Renal effects of uric acid: hyperuricemia and hypouricemia
Article information

Abstract
The prevalence of chronic kidney disease (CKD) is increasing worldwide. Although hyperuricemia has been associated with CKD in many studies, it remains controversial whether this is the cause or the result of decreased renal function. Recent observational studies of healthy populations and patients with CKD have reported that uric acid (UA) has an independent role in the development or progression of CKD. Experimental studies have shown several potential mechanisms by which hyperuricemia may cause or promote CKD. However, other reports have indicated an association between hypouricemia and CKD. This opposing effect is hypothesized to occur because UA is a major antioxidant in human plasma and is associated with oxidative stress. In this article, we discuss the potential association between UA imbalance and CKD and how they can be treated.
INTRODUCTION
Chronic kidney disease (CKD) is a significant public health issue globally, with an estimated prevalence of 7.9% in Korea [1]. The number of patients undergoing dialysis in Korea has continued to increase due to worsening kidney function in patients with CKD [2]. CKD is not only associated with developing end-stage renal disease (ESRD) but also with an increased risk of cardiovascular disease (CVD) [3]. Indeed, the hypothesis that uric acid (UA) could mediate CVD or hypertension was proposed nearly 140 years ago [4]. In 1889, it was proposed that UA mediated diseases, such as CKD, hypertension, and diabetes [5], and in 1897, hyperuricemia and gout were reported to be associated with renal dysfunction [6]. According to data collected when no drugs were available to reduce serum UA levels, proteinuria was common (occurring in 25% of cases) in patients with gout, renal function frequently decreased (50% of cases), and ESRD occurred in 10% to 25% [7,8]. Although evidence shows that UA is associated with the development of CKD, controversy remains as to whether this is the cause or the result of decreased renal function [9,10]. Most studies have shown a relationship between CKD and hyperuricemia, but some have also shown an association between decreased renal function and hypouricemia [11,12].
URIC ACID METABOLISM
UA is generated in the liver from the catabolism of exogenous and endogenous purine mononucleotides into hypoxanthine and guanine [13]. Hypoxanthine is further oxidized by xanthine dehydrogenase/oxidase to form xanthine, which is further oxidized by the same enzyme to form UA. This constitutes the end product of purine nucleotide metabolism in higher primates [14]. In other mammals, UA is further degraded by uricase, an enzyme that primates lack, to become allantoin [15], which is more water-soluble than UA and can be efficiently excreted in urine [16]. Mammals with the uricase enzyme usually show lower UA levels (1 to 2 mg/dL), whereas primates have UA levels that are 3- to 10-times higher [17].
In humans, approximately two-thirds of all UA is excreted in the urine, and one-third is excreted in the gastrointestinal tract [18]. UA is filtered by the glomeruli and reabsorbed by the proximal tubule with a normal fractional excretion of 10% [13]. Urate transporter 1 (URAT1; SLC22A12) reabsorbs glomerular-filtrated UA and is localized on the luminal side of proximal tubule cells [19], while glucose transporter 9 (GLUT9; SLC2A9) allows intracellular UA to exit through the basolateral side of the cells (Table 1) [18,20]. Otherwise, UA secretion is mediated by ATP-binding cassette transporter subfamily G member 2 (ABCG2) and sodium-dependent phosphate transporter 1 (NPT1; SLC17A1) and 4 (NPT4; SLC17A3) [21-23]. Studies of polymorphisms in these transporters suggest that renal overload hyperuricemia is a novel pathophysiological mechanism in gout [24]. However, in an experimental study of 5/6 nephrectomized rats, the expression levels of URAT1, GLUT9, ABCG2, and NPT4 in the proximal tubules decreased, which may have been associated with CKD-related tubular injury [25]. This suggests that tubular urate transporters do not cause hyperuricemia directly; instead, this results from a decrease in UA filtered by the glomeruli [26].
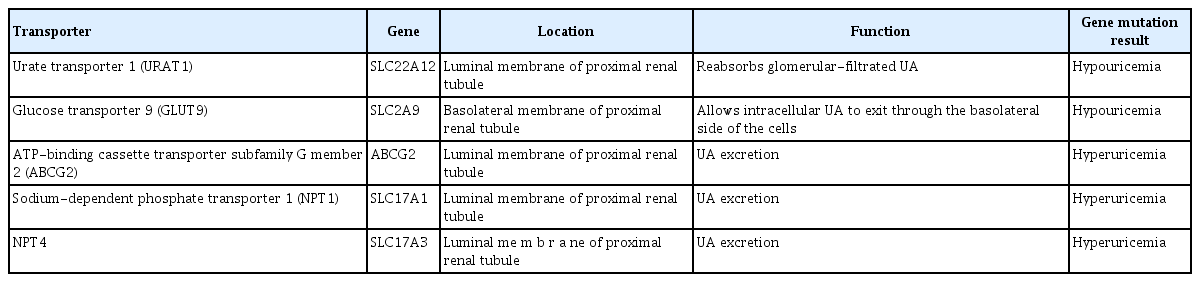
Renal urate transporters and associated conditions
Mechanisms of hyperuricemia
Hyperuricemia has been defined as ≥ 7.0 mg/dL in men, and ≥ 5.7 [27] or ≥ 6 mg/dL [28] in women. Although hyperuricemia occurs in patients with CKD, the fact that hyperuricemia can precede CKD suggests that other factors are relevant. Risk factors for CKD include metabolic syndrome, which is associated with hyperuricemia [29], insulin resistance and hyperinsulinemia, which reduce urinary UA excretion [30], and hypertension, which increases renal vascular resistance and UA retention [31]. However, recent studies have shown that hyperuricemia, i.e., increased serum UA levels, may prevail in these conditions and occur earlier, indicating that other factors may be the underlying cause of hyperuricemia [32,33]. For example, diets high in meat, seafood, sugar, and beer are all associated with an increased risk of hyperuricemia, whereas diets rich in dairy products are associated with a decreased risk [34]. Children with a history of low birth weight also show increased serum UA levels, endothelial dysfunction, and hypertension [35]. Finally, genetic factors are also involved. Familial juvenile hyperuricemic nephropathy is a rare autosomal dominant disease that results from a mutation in the uromodulin gene. It is characterized by abnormal handling of UA and hyperuricemia, and it is often complicated by gouty arthritis, with renal failure occurring due to tubulointerstitial nephritis [36]. In some studies, plasma volume contraction and compensatory higher reabsorption activity of the proximal tubule including upregulation of Na+-coupled urate transporters was suggested to be the cause of the hyperuricemia in cases of the uromodulin gene mutation [37,38]. A genome-wide association study of gout and its subtypes was performed to identify novel gout loci [39].
Mechanisms of hypouricemia
Hypouricemia is defined as a serum UA of ≤ 2.0 mg/dL [40]. In a Japanese population, it was reported to occur in 193 of 90,710 men (0.2%) and 540 of 136,935 women (0.4%) [11], while in Korea, the prevalence has been reported to be 4.14% (299/7,223) among inpatients and 0.53% (125/23,534) among outpatients, with an overall prevalence of 1.39% (424/30,757) [41]. Hypouricemia can be divided into conditions that result from decreased UA production and those that result from increased renal clearance.
Hereditary xanthinuria is an autosomal recessive disease caused by a deficiency of the xanthine dehydrogenase/oxidase enzyme that results in decreased UA production. This disorder is marked by hypouricemia, low urinary concentrations of urate, and high urinary concentrations of xanthine, which lead to the development of urinary xanthine kidney stones [42].
Urate-lowering drugs, such as allopurinol and febuxostat, inhibit xanthine oxidase, and cause hypouricemia. Pegloticase, a mammalian recombinant pegylated uricase enzyme that converts urate into allantoin has 5 to 10-fold increased solubility. When used as indicated to treat severe refractory gout it converts urate into the more soluble allantoin that is more readily excreted by the kidney. Using pegloticase can cause severe hypouricemia with UA levels falling below 2 mg/dL in some patients [43,44]. Several other uricosuric agents also decrease serum UA levels [45]. These include probenecid, benzbromarone, angiotensin II receptor blockers, fenofibrate, trimethoprim-sulfamethoxazole, and high-dose salicylate [46-48].
Gene mutations can also cause renal hypouricemia, such as those of SLC22A12, encoding URAT1 [49-53], and SLC2A9, encoding GLUT9 [54,55]. Most patients are asymptomatic, but some may be predisposed to urinary tract stones or exercise-induced acute renal failure [56]. In the latter case, patients will show a high fractional excretion of UA (> 10%).
Hypouricemia has also been reported in patients with diabetes [57,58], where it is known to occur due to increased renal excretion and only in patients with normal renal function [59,60]. Low levels of serum UA may also develop secondary to expansion of extracellular volume, which reduces proximal reabsorption of sodium and UA [61]. This is common when patients receive large volumes of intravenous fluid or when patients have psychogenic polydipsia or syndrome of inappropriate antidiuretic hormone. Several hepatic diseases, such as cholangiocarcinoma, viral hepatitis, and primary biliary cirrhosis, have been associated with hypouricemia due to abnormal UA reabsorption [62-64]. Patients with Hodgkin’s disease may also develop marked hypouricemia because of an isolated defect in urate reabsorption [65], though successful treatment of the underlying disease appears to correct this abnormal renal handling of urate.
PATHOPHYSIOLOGY OF URIC ACID-RELATED KIDNEY DISEASE
Hyperuricemia and kidney
Kidney damage due to hyperuricemia has traditionally been thought to result from the effect of UA crystals [66]. Hyperuricemia associated with hyperuricosuria (urinary UA excretion > 800 mg/day in men and > 750 mg/day in women) has been postulated to cause acute kidney damage by depositing crystals intraluminally in the collecting ducts. In turn, this causes tubular obstruction, the development of an inflammatory response, and progressive tubulointerstitial damage over time that lowers the estimated glomerular filtration rate (eGFR). Multiple crystal-independent mechanisms have been suggested in basic research, including renal vasoconstriction mediated by endothelial dysfunction, activation of the renin-angiotensin system, afferent arteriolopathy, and the epithelial-to-mesenchymal transition in renal tubular cells [67-72].
It is assumed that UA acts as a powerful antioxidant in the extracellular environment, but that it is a prooxidant inside the cell, where it stimulates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [72]. UA has been reported to induce endothelial dysfunction by increasing oxidative stress and decreasing the bioavailability of endothelial nitric oxide [73]. Some research suggests that lowering UA with allopurinol may improve endothelial dysfunction in humans [74,75].
In animal models, hyperuricemia increases cyclooxygenase-2 expression and leads to the proliferation of vascular smooth muscle cells in preglomerular arterioles [70]. Hyperuricemia in rats induces hypertension, renal injury, and fibrosis, in part by activating the renin-angiotensin system [67]. Preglomerular vasculopathy caused by hyperuricemia deranges the autoregulatory response of afferent arterioles and triggers glomerular hypertension [69,76]. In addition, lumen obliteration induced by vascular wall thickening results in severe vasoconstriction that decreases renal blood flow, the eGFR, and perfusion to peritubular capillaries, causing tubulointerstitial inflammation, tubulointerstitial fibrosis, and arterial hypertension [71]. UA induces tubulointerstitial fibrosis via the epithelial-to-mesenchymal transition in renal tubular cells [77].
Hypouricemia and kidney
UA is an important antioxidant in human plasma and, as such, hypouricemia has been proposed to increase the risk of a decline in kidney function by reducing antioxidant capacity [78-80]. Consistent with this, hypouricemia has been associated with several inflammatory and degenerative diseases, including acute graft-versushost disease, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, and multiple sclerosis [81,82]. These associations have been attributed to reduced antioxidative capacity [81,82].
Hypouricemia has also been associated with urolithiasis and exercise-induced acute kidney injury (EIAKI), particularly in patients with hereditary hypouricemia [83,84]. Because UA acts as an antioxidant that protects endothelial function, hypouricemia causes EIAKI through renal artery spasm, with a clinical picture that is characterized by nausea, vomiting, and loin, and abdominal pain. In a Japanese study, all patients with EIAKI later recovered their kidney function, but 24% experienced recurrent acute kidney injury (AKI) [85]. Pathology revealed chronic lesions, such as thickening of the tubular basement membrane and interstitial fibrosis, despite a normal creatinine clearance rate in some patients who experienced recurrent AKI. Although the loss to follow-up meant that the researchers did not report if these patients developed CKD, it is certainly plausible that recurrent AKI can lead to CKD [86]. In addition, excessive urinary excretion of UA can result in the formation of UA crystals, causing urolithiasis in patients with hypouricemia [87]. Urolithiasis and EIAKI have a prevalence of 8.5% and 6.5%, respectively, in patients with hereditary renal hypouricemia based on genetic testing [51].
RELATIONSHIP BETWEEN URIC ACID AND CKD PROGRESSION: OBSERVATIONAL STUDIES
Hyperuricemia and CKD
Many large observational studies have examined the association between serum UA and the development or progression of CKD in both the general population and in patients with existing CKD (Table 2) [88-99]. Most observational studies have shown positive results, with hyperuricemia being an independent risk factor for the development and progression of kidney disease in diabetic or non-diabetic patients [88-95,99,100]. In a meta-analysis involving 13 observational trials of 190,718 patients with normal renal function, hyperuricemia was an independent risk factor for developing CKD [100]. The summary odds ratio (OR) for the association between hyperuricemia and developing new-onset CKD increased with increasing follow-up, indicating that hyperuricemia may play a role in the long-term progression of renal function. A subgroup analysis revealed a stronger association between hyperuricemia and CKD development in Western than in Asian populations. This difference may result from racial, geographic, or dietary differences because the Western diet possibly contains more purine-rich foods.
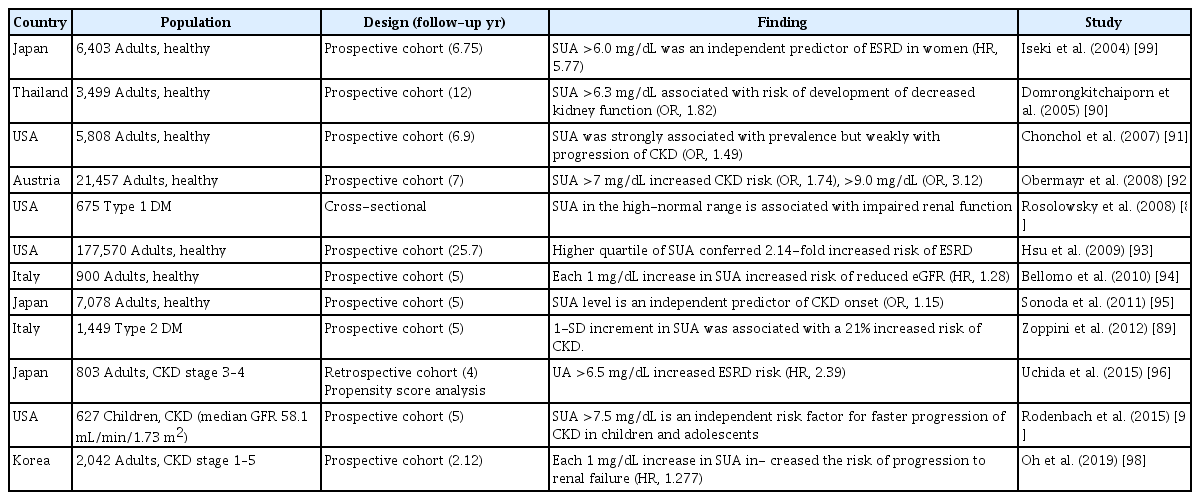
Observational studies of hyperuricemia and CKD
Serum UA in the high-normal range (≤ 4.0 mg/dL vs. > 4.0 mg/dL) is associated with impaired renal function in patients with type 1 diabetes [88]. In people with type 2 diabetes and preserved kidney function, hyperuricemia (≥ 7.0 mg/dL in men and ≥ 6.5 mg/dL in women or those on allopurinol therapy) is an independent risk factor for incident cases of CKD [89]. In a Japanese screened cohort study, hyperuricemia (serum UA > 6.0 mg/dL) was an independent predictor of ESRD in women [99], but there was no significant association in men. A previous study in which ESRD was followed up for 25 years reported hyperuricemia as an independent risk factor for ESRD [93], and in a prospective follow-up study of 21,475 healthy participants, hyperuricemia independently increased the risk for new-onset kidney disease [92].
Understanding the role of UA in predicting the progression of kidney disease in patients with CKD is more controversial due to the unavoidable causal relationship. Uchida et al. [96] used a propensity score analysis to clarify the independent effect of UA on the subsequent risk of ESRD. They showed that higher serum UA accelerated the progression to subsequent ESRD in a retrospective CKD cohort. In another study of children and adolescents with a median age of 12.3 years, hyperuricemia was an independent risk factor for faster CKD progression [97]. In a Korean cohort study of patients with CKD and a mean eGFR of 52.80 ± 12.3 mL/min/1.73 m2, the risk of progression to renal failure increased by 28% (hazard ratio [HR], 1.277; 95% confidence interval [CI], 1.212 to 1.345) for each 1 mg/dL increase in the baseline UA level [98]. Multivariate analysis found an association between the upper UA quartile and an increased risk of composite renal outcome (HR, 3.590; 95% CI, 2.546 to 5.063). Hyperuricemia was associated with the development of CKD in a risk score model [101,102]. Hyperuricemia is associated with sarcopenia, and sarcopenia is associated with CKD progression, death, and CVD in CKD patients [103,104]. Fat accumulation in and around the kidneys is associated with hyperuricemia and the development of CKD [105,106].
Other observational studies have failed to show a significant relationship between hyperuricemia and the progression of CKD [107,108]. In a study of patients with mild-to-moderate CKD and no diabetes, increasing serum UA levels predicted disease progression only when the analysis was not adjusted for baseline kidney function parameters. After adjusting for baseline eGFR and proteinuria, the association completely vanished [107]. Similarly, a analysis of the Modification of Diet in Renal Disease study, which enrolled 838 patients with stage 3 to 4 CKD, showed that hyperuricemia was an independent risk factor for all-cause and CVD mortality, but not kidney failure [108]. Serum UA levels of 3,885 patients with stage 2 to 4 CKD in the Chronic Renal Insufficiency Cohort were examined, and serum UA levels were reported to be an independent risk factor for progression to dialysis or transplantation when the initial eGFR was > 45 mL/min/1.73 m2 but not when it was < 30 mL/min/1.73 m2 [109]. The authors postulated that higher UA levels at preserved eGFRs have more relevance for kidney failure than at lower GFRs. When GFR is preserved, the deleterious effects of UA may therefore be more pathogenic and easier to discern than when kidney function is worse, at which point factors that govern the increase in UA levels and increased morbidity may be more important.
Hypouricemia and CKD
A U-shaped association has been reported between serum UA level and CVD mortality, suggesting that both hyperuricemia and hypouricemia are risk factors for CVD mortality [110,111]. However, there are few reports of this association between serum UA and the loss of kidney function.
In a Japanese prospective cohort study among healthy people, both decreased and increased serum UA levels were associated with a loss of kidney function [112]. The study population comprised 104,796 asymptomatic people. Of the 9,847 without CKD, 4,188 were followed up for at least 3 years, 3,102 for 6 years, and 1,052 for 9 years. Overall, the mean serum UA level was 5.8 ± 1.2 mg/dL in men and 4.1 ± 0.9 mg/dL in women. Many men had incident CKD and a > 25% decrease in eGFR in groups 4 (serum UA ≥ 6.5 mg/dL) and 1 (serum UA ≤ 5.0 mg/dL), respectively. In subjects with low serum UA levels (men, < 5 mg/dL; women, < 3.6 mg/dL), multivariate linear mixed models showed a time-dependent association with declining eGFR. Logistic additive models also showed that both high and low serum UA levels affected the likelihood of the outcome events more than intermediate levels among men, but not among women.
In a population-based cross-sectional study, hypouricemia (serum UA ≤ 2 mg/dL) was associated with reduced kidney function in men (OR, 1.83; 95% CI, 1.23 to 2.74), but not in women (OR, 0.61; 95% CI, 0.43 to 0.86), when compared against a reference category (serum UA levels of 4.1 to 5.0 mg/dL) and after adjusting for age, drinking, smoking, diabetes, hypertension, hypercholesterolemia, obesity, and history of renal failure [11]. In another Japanese cross-sectional study in the general population, the rates of previous urinary stones and kidney diseases (including nephritis/nephrosis) were 1.2% (3.3% men, 0.7% women) and 2.3% (10% men, 0.7% women), respectively. Men with hypouricemia had a 9-fold higher rate of previous kidney diseases compared with those who had no hypouricemia [113].
Finally, a Korean prospective cohort study in a rural population revealed that both high and low serum UA levels were risk factors for the development of CKD in men, but that only high levels were a risk factor in women [114]. Among the 5,577 participants, 9.4% of men and 11.0% of women developed CKD. The HR for CKD was higher in the fourth quintile of serum UA levels than in the third quintile in men (adjusted HR, 1.60; 95% CI, 1.02 to 2.51) and women (adjusted HR, 1.56; 95% CI, 1.14 to 2.15). The development of CKD was also more common in the lowest quintile of serum UA levels than in the third quintile in men (adjusted HR, 1.83; 95% CI, 1.15 to 2.90). Patients with ESRD and low serum UA levels were also reported to have a higher mortality risk than those with high serum UA levels [115].
EFFECTS OF URIC ACID-LOWERING THERAPY ON CKD
In a meta-analysis of 12 studies, Liu et al. [116] reported that UA-lowering therapy is associated with slowed CKD progression. The pooled estimate for eGFR favored UA-lowering therapy with a mean difference of 3.88 mL/min/1.73 m2 (95% CI, 1.26 to 6.49). The risk of worsened kidney function, ESRD, or death decreased significantly in the treatment group compared with the control group (relative risk, 0.39; 95% CI, 0.28 to 0.52) [116]. Clinical trials of UA-lowering therapy published to date are listed in Table 3 [117-124]. In these studies, UA levels during treatment remained at 5 to 6 mg/dL.

Clinical trials of UA-lowering therapy on CKD
Allopurinol
Allopurinol is metabolized by xanthine oxidase to oxypurinol, which in turn, noncompetitively inhibits xanthine oxidase [13]. Allopurinol is usually started at 100 mg daily in patients with normal renal function and titrated every 2 to 4 weeks to the minimum dose required to achieve and maintain the goal range of serum UA. The half-lives of allopurinol and its active metabolite, oxypurinol, are prolonged in patients with renal dysfunction [125]. A daily starting dose of allopurinol of < 1.5 mg/mL/min of eGFR is advised in such patients, with dose increments of no more than 50 mg daily every 4 weeks to the minimum daily dose necessary to achieve the goal [126,127]. Among relatively mild adverse reactions occurring in 3% to 5% of patients treated with allopurinol are rash, leukopenia or thrombocytopenia, and diarrhea. Severe reactions, such as severe cutaneous adverse reactions, may occur very rarely, but still occur in subjects with the human leukocyte antigen (HLA) B*58:01 allele, CKD, and use of a thiazide/loop diuretic [128,129].
Several studies have investigated the effect of allopurinol therapy on kidney outcomes [117,118,120,130]. Siu et al. [117] reported that it slowed renal disease progression in hyperuricemic subjects with mild-to-moderate CKD (1.35 < serum creatinine ≤ 4.50 mg/dL) after 1 year. In a prospective controlled study, Goicoechea et al. [118] further reported that allopurinol preserved renal function. They randomly assigned patients with CKD either to receive allopurinol 100 mg/day or to continue with their usual therapy. A decrease of the serum UA level from 7.8 ± 2.1 to 6.0 ± 1.2 mg/dL was not associated with a significant change in the eGFR (40.8 ± 11.2 to 42.2 ± 13.2 mL/min/1.73 m2), whereas the eGFR fell in the control group (39.5 ± 12.4 to 35.9 ± 12.3 mL/min/1.73 m2). Allopurinol treatment was also associated with fewer CVD events (seven in the allopurinol group and 15 in the control group). Momeni et al. [119] reported that allopurinol therapy reduces proteinuria in patients with diabetic nephropathy, but with no effect on creatinine.
It is certainly possible that the favorable results associated with allopurinol are due to effects other than its UA-lowering effects. For example, allopurinol blocks the production of reactive oxygen species, key signaling molecules in inflammatory diseases [131], which are generated during the conversion of xanthine to UA [9]. Other potential mechanisms include adenosine accumulation, which is anti-inflammatory and inhibits tumor necrosis factor-α, nuclear factor kappa-light-chain-enhancer of activated B cells, and the NLRP3 inflammasome (i.e., NACHT, LRR, and PYD domain-containing protein 3) [132]. Furthermore, allopurinol treatment improves peripheral and cerebrovascular endothelial function [74,133-136]. These mechanisms may each be associated with kidney protective effects either alone or in combination.
Febuxostat
Febuxostat is a relatively new xanthine oxidase inhibitor that is safe for patients with kidney dysfunction [137,138]. Febuxostat is used to treat hyperuricemia in gout patients at daily doses of 40 and 80 mg [139]. Several types of adverse effects have been associated with the use of febuxostat, some of which include cardiovascular and hepatic abnormalities that may be more common with febuxostat than with allopurinol [140].
In a randomized controlled trial of 93 patients with stage 3 and 4 CKD, febuxostat slowed a decline in eGFR compared to placebo [121]. The mean eGFR in the febuxostat group increased nonsignificantly from a baseline of 31.5 ± 13.6 to 34.7 ± 18.1 mL/min/1.73 m2 at 6 months; with placebo, the mean eGFR decreased from 32.6 ± 11.6 to 28.2 ± 11.5 mL/min/1.73 m2. In total, 17 of the 45 (38%) participants in the febuxostat group showed a 10% decline in eGFR compared with 26 of 48 (54%) in the placebo group. However, a larger randomized controlled trial failed to show a beneficial effect [122], with no significant difference in mean eGFR slopes between the febuxostat (0.23 ± 5.26 mL/min/1.73 m2 per year) and placebo (−0.47 ± 4.48 mL/min/1.73 m2 per year) groups (difference, 0.70; 95% CI, −0.21 to 1.62; p = 0.10).
Several other recent studies have compared febuxostat to placebo or allopurinol, and febuxostat retarded the decline in renal function more effectively than allopurinol [116,123,124]. In a Korean retrospective study, a febuxostat group had significantly lower mean serum UA levels (5.7 ± 1.0 mg/dL) than either an allopurinol group (7.1 ± 1.2 mg/dL) or a control group, (8.0 ± 0.8 mg/dL) (p < 0.001) and maintained significantly higher mean eGFR values for 4 years. The febuxostat group also had significantly longer survival times free from renal disease progression (87.7 months; 95% CI, 71.2 to 104.2) compared with either the allopurinol group (77.6 months; 95% CI, 60.2 to 94.9) or the control group (48.7 months; 95% CI, 39.3 to 58.1) (p < 0.001) [123]. In a Chinese prospective cohort study of patients with stage 3 to 5 CKD, the serum UA target of < 6.0 mg/dL was achieved by 96.4% of patients in the febuxostat group and by 37.5% in the allopurinol group at 6 months. The eGFR in the febuxostat group increased from 28.45 to 30.65 mL/min/1.73 m2 at 6 months, while it decreased from 28.06 to 24.39 mL/min/1.73 m2 in the allopurinol group [116]. In another Chinese prospective cohort study of patients with stage 2 to 3 CKD, the proportion of patients showing a≥ 10% decline in eGFR from baseline decreased by 17.9% in the febuxostat group and 34.1% in the allopurinol group (p = 0.025) [124].
CONCLUSIONS
A 100-year history of basic and clinical studies supports UA being a direct risk factor for CKD. Nevertheless, controversy exists over the causal role of UA in the development or exacerbation of CKD, with conflicting results being produced in various studies. Although many nephrologists currently treat asymptomatic patients with hyperuricemia [141], there is no consensus on the appropriateness of such a treatment approach [142]. Furthermore, hypouricemia has been shown to increase the risk for deterioration of renal function, making it more difficult to set a target range for the optimal serum UA level. In the CARES trial, all-cause and cardiovascular mortality were higher in the febuxostat group than in the allopurinol group, even though patients receiving febuxostat had lower serum UA levels [140]. The higher mortality associated with this more intense UA-lowering therapy is consistent with the U-shaped association between UA and mortality proposed in some observational studies [110,111]. Because UA is the most abundant antioxidant in plasma, strategies to increase UA are ongoing in clinical trials of patients with neurological diseases [143,144]. Further research is needed to assess the safety of lowering serum UA to specific thresholds to produce safe guidelines [145]. Optimal serum UA levels should be defined at both upper and lower limits, for men and women, and in patients with and without CVD or CKD.
Notes
No potential conflict of interest relevant to this article was reported.