The clinical impact of gut microbiota in chronic kidney disease
Article information

Abstract
Gut microorganisms play critical roles in both maintaining host homeostasis and the development of diverse diseases. Gut dysbiosis, an alteration of the composition and function of gut microorganisms, is commonly seen in patients with chronic kidney disease (CKD). CKD itself contributes to a disruption of the symbiotic relationship between the gut microbiota and the host, while the resulting gut dysbiosis may play a part in stage progression of CKD. This bidirectional relationship supports the concept that the gut microbiota is considered a novel focus for the pathogenesis and management of CKD. This article examines the interaction between the gut microbiota and the kidney, the mutual effects of dysbiosis and CKD, and possible treatment options to restore gut eubiosis, and reduce CKD progression and its related complications.
INTRODUCTION
Chronic kidney disease (CKD) is a global health threat with a prevalence of 8% to 16% worldwide, and has been gradually increasing in recent decades due to the aging population, the increase in chronic debilitating diseases, and decreased mortality of patients at risk for renal impairment [1]. CKD is mainly caused by diabetes mellitus (DM), hypertension (HTN), and glomerulonephritis, eventually progresses to end-stage renal disease (ESRD) and is associated with high morbidity and mortality related to cardiovascular diseases, despite active medical treatment [1-3]. Regardless of attempts to attenuate the progression of CKD, there have been no significant breakthroughs in the management of patients with CKD for decades, due to a lack of understanding of the pathogenesis and pathophysiology of CKD. Recently, as interest in the gut microenvironment has increased, the gut microbiota is seen as a key modulator of human health, to the extent that it has been proposed to be an essential organ in the human body [4,5]. The relationship between the gut microbiota and the host organs has attracted great attention in both the maintenance of host homeostasis and the development of diverse diseases [4-7]. CKD contributes to the alteration of composition and function of the gut microbiota, called dysbiosis. In contrast, there is growing evidence that dysbiosis itself contributes to CKD development and progression via several mechanisms such as microbiota-derived toxins, immune-mediated products and neuroendocrine-mediated substances [6-10]. This bidirectional relationship supports the concept that the gut microbiota is considered a novel focus for the cause and treatment of CKD. This review will explore the interface between the gut microbiota and kidney, address the clinical effects of gut microbiota on CKD onset and progression, and include the results of clinical trials for possible treatment options in patients with CKD.
SYMBIOSIS: GUT MICROBIOTA AND HEALTHY SUBJECTS
The gut microbiota is defined as all the living gut microorganisms that form a symbiotic relationship with the host, and includes mainly bacteria, but also viruses, archaea, fungi, and unicellular eukaryotes [4,5]. The human gut contains approximately 1 trillion microorganisms with thousands of species encoding more than 3 million genes (150-fold more than the human genome) [4]. In healthy subjects, the microbiota in the colon is mainly composed of five phyla; significant proportions are Firmicutes and Bacteroidetes (collectively 90%), followed by Actinobacteria, Verrucomicrobia, and, in a lower proportion, Proteobacteria [4,5,10]. The microbiota plays a fundamental role in the breakdown of indigestible plant polysaccharides from the diet, biosynthesis of short-chain free fatty acids (SCFAs) and vitamins, biotransformation of conjugated bile acids and degradation of dietary oxalates, induction and training of our immune system, and reducing allergic responses to foods and/or environmental antigens [4-8]. The colonization of microbiota in the gut begins immediately after birth and reaches a mature composition by the age of 3 years. Thereafter, it is maintained throughout adulthood. However, the pattern of microbiota including its richness, diversity, and uniformity, is influenced by age, hygiene, infection, exposure to allergens, drugs, and several diseases [9,10]. Therefore, an imbalance in composition of the gut microbiota may be related to disease development.
THE EFFECT OF CKD ON GUT MICROBIOTA
CKD is characterized by the accumulation of uremic toxins, and is commonly associated with disturbed mechanical changes in the gastrointestinal tract such as edema of the intestinal wall and alteration of colonic transit [8,9,11]. Patients with CKD often experience dietary restrictions and the frequent consumption of iron, phosphorus binding agents, and antibiotics [8-12]. These characteristics may induce the development of dysbiosis, and furthermore may inversely promote disease progression to advanced stages of CKD. Fig. 1 shows a reciprocal interaction between gut dysbiosis and CKD.

The reciprocal interaction between gut dysbiosis and CKD. UT, uremic toxin; LPS, lipopolysaccharide; SCFA, short-chain free fatty acid; IS, indoxyl sulfate; pCS, p-cresyl sulfate; TMAO, trimethylamine N-oxide; TLR4, toll-like receptor-4; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α; ROS, reactive oxygen species; GLP-1, glucagon-like peptide 1; PYY, peptide YY; RAAS, renin-angiotensin-aldosterone system; GABA, gamma aminobutyric acid; ACh, acetylcholine; CKD, chronic kidney disease; MBD, mineral bone disorder; PTH, parathyroid hormone.
The effect of uremic toxins from CKD on dysbiosis
As renal function decreases, waste products called uremic toxins accumulate in the blood, and also increase in the intestinal epithelium [8,9,13]. This change in the gut promotes the colonization of bacteria that can use urea as an energy source by producing the enzymes urease or uricase. Urease is a cytosolic enzyme that catalyzes the hydrolysis of urea to ammonia and carbon dioxide. Consequently, the markedly increased formation of ammonia increases the gut pH, mediates enterocolitis by the breakdown of gut epithelial tight junctions, and facilitates endotoxemia and systemic inflammation [7-9,13-16]. The uric acid originating from purine metabolism is mainly excreted via the kidney; however as renal function decreases, the colon replaces the kidney as the primary site of uric acid excretion [9,15,16]. The high concentration of uric acid increases the bacteria containing urease and uricase and influences changes in the gut microenvironment via the same pathway. Several studies have demonstrated that CKD patients have significant changes in the gut microbiota with loss of α-diversity, β-diversity, and richness [8-10]. In a study of 24 stable ESRD patients, Wong et al. [17] also showed reduction in the Lactobacillaceae and Prevotellaceae families, which can express the butyrate kinase gene involved in protective processes, and confirmed that most of the microbiota has urease, uricase or tryptophanase activity; 12 of 19 bacteria families have urease activity (including Alteromonadaceae, Cellulomonadaceae, Clostridiaceae), five have uricase activity (Cellulomonadaceae, Dermabacteraceae, Micrococcaceae, Polyangiaceae, and Xanthomonadaceae), and three have tryptophanase activity (Clostridiaceae, Enterobacteriaceae, and Verrucomicrobiaceae). This alteration of gut microbiota can promote the acceleration of dysbiosis and its metabolic activities, and lead to the change of gut structure called leaky gut syndrome. Vaziri et al. [16] reported a marked reduction in the amount of tight junction-related proteins such as claudin-1, occludin, and zonula occludens 1 in the colonic mucosa of animals with CKD. In addition, Farhadi et al. [18] showed several histological changes such as reduction of villous height, elongation of the crypts, and infiltration of the lamina propria with inflammatory cells in the intestines of CKD patients. These pathological changes in the gut suggest that translocation of bacteria and the influx of endotoxins across the intestinal wall can contribute to inducing systemic inflammation in CKD.
Other CKD-related factors that contribute to dysbiosis
The classic CKD diet is generally composed of light meals containing low concentrations of sodium, potassium and phosphate, to reduce their adverse metabolic effects [19]. This type of diet limits the consumption of potassium-rich foods such as fruits, vegetables, nuts, and high-fiber products. Indigestible carbohydrates are not only essential nutrients for the metabolism of gut microbiota but also precursors of short-chain fatty acids (SCFAs) related to immune regulation, glucose and lipid metabolism, gut integrity, and appetite [4,19,20]. Therefore, the CKD diet contributes to a decrease in the production of SCFAs and increases the amino nitrogen load, which can transform into uremic toxins [8,20]. CKD patients are commonly prescribed multiple drugs including iron-containing compounds, phosphate binders, and antibiotics [20,21]. This long-term and high pill burden for CKD patients can extensively influence the composition and richness of the gut microbiota.
THE EFFECT OF DYSBIOSIS ON CKD PROGRESSION
CKD-induced dysbiosis can contribute to CKD progression and the development of CKD-related adverse events through diverse mechanisms such as microbiota-derived metabolites, a disrupted intestinal barrier, and changes in the neuroendocrine immune system.
The effect of microbiota-derived toxins on CKD
Uremic toxins can be classified based on their site of origin: endogenous (mammalian metabolism), exogenous (diet), or microbial [22,23]. Among these original site-specific toxins, three molecules including indoxyl sulfate (IS), p-cresyl sulfate (pCS), and trimethylamine N-oxide (TMAO), are microbiota-derived uremic toxins involved in the development of complications including cardiovascular disease and mortality as well as the pathogenesis and progression of CKD. IS, a protein-binding uremic toxin and one of the microbiota-derived metabolites, is synthesized from dietary tryptophan by bacterial fermentation, and is excreted in the urine. IS converted from indole in the liver contributes to peripheral vascular disease and thrombosis of vascular access [22-24]. Its serum concentration increases in proportion to a decrease in renal function. In a prospective study of 268 CKD patients [24], Wu et al. [24] reported that the baseline concentration of IS can be a predictor of loss of renal function. Some experimental studies have demonstrated that IS mediates the renal expression of genes related to tubulointerstitial fibrosis, such as transforming growth factor β1 and a tissue inhibitor of metalloproteinases [23,25]. Another experimental study showed that mouse podocytes exposed to IS for 8 weeks exhibited a pro-inflammatory phenotype, perturbed actin cytoskeleton, decreased expression of podocyte-specific genes, and decreased cell viability [26]. pCS is a colonic fermentation product of tyrosine and phenylalanine catabolism by anaerobic gut bacteria. Once p-cresol is absorbed, it is conjugated with other substances in the liver, where a sulfate group is added [6-8]. In a CKD rat model, pCS increased the production of reactive oxygen species (ROS), activating nicotinamide adenine dinucleotide phosphate oxidase and increasing caspase-3 activity, leading to an increased apoptosis ratio [27]. In another experimental study of half-nephrectomized mice, IS or pCS activated the intrarenal renin-angiotensin-aldosterone system (RAAS) and induced interstitial fibrosis and glomerulosclerosis [28]. TMAO is synthesized from dietary choline, phosphatidylcholine, and L-carnitine. Pelletier et al. [29] reported that the serum TMAO concentration negatively correlates with the glomerular filtration rate (GFR) in patients with CKD. Tang et al. [30] showed that an elevated level of TMAO was associated with the degree of tubulointerstitial fibrosis in an animal model and poorer overall survival in CKD patients. Stubbs et al. [31] also reported that TMAO increases the phosphorylation of SMAD3, an important regulator of fibrosis, and consequently enhances atherosclerosis and thrombosis, leading to an increased incidence of coronary artery disease. Therefore, TMAO has been proposed as a potential surrogate marker to detect early cardiovascular risk in patients with CKD. These accumulating data suggest that increased microbiota-derived metabolites are definite risk factors that contribute to CKD development and progression, and attempting to reduce microbiota-derived uremic toxins seems to be a reasonable therapeutic strategy for CKD patients.
The immunological effects of dysbiosis on CKD
The gut microbiota can provide a large number of potential immunostimulatory bacterial products. Recently, this has been considered a critical exogenous trigger of host immune dysfunction [4,5]. The leaky gut in CKD patients can facilitate the influx of microbiota-derived products into the hepatic portal system and systemic circulation across the intestine wall. A lipopolysaccharide (LPS) is derived from the cell wall component of gram-negative bacteria [32-34]. LPS induces systemic inflammation via a cascade of inflammatory responses. Lipid A is a component of LPS and serves as the microbe-specific molecular signal that binds to the surface receptor complexes of immune cells (monocytes/macrophages), which comprise toll-like receptor-4 (TLR4) and myeloid differentiation (MD) factor-2. The formation of the TLR4-MD2-LPS complex activates the signaling pathway controlling the expression of inflammatory genes, leading to excessive production of pro-inflammatory cytokines such as interleukin-1β (IL-1β), IL-6, and tumor necrosis factor α [33-35]. TLR4 is also highly expressed in the endothelial cell, vascular smooth muscle cell and adventitial fibroblast. Activated TLR4 promotes the recruitment of monocytes, transformation of macrophages to foam cells, and the expression of adhesion molecules, ROS, and pro-coagulant activity by intracellular inflammatory pathways mediated through nuclear factor-κB and mitogen-activated protein (MAP) kinases [32-35]. Ultimately, these processes can initiate and promote atherosclerosis. McIntyre et al. [36] demonstrated that circulating bacterial endotoxin/LPS levels are increased in all stages of CKD and reach the maximum in dialysis patients, and suggested that systemic inflammation is also a strong and independent predictor of mortality in CKD. Szeto et al. [32] also showed that circulating endotoxin in patients undergoing peritoneal dialysis is related to systemic inflammation and features of atherosclerosis.
The neuroendocrine effects of dysbiosis on CKD
The gut is the second-most innervated organ in the body, and the gut microbiota communicates with the nervous system by producing several hormones and neurotransmitters [4,7]. The central and peripheral nervous systems regulate the circulatory system by modulating the sympathetic nervous system, RAAS, and pituitary hormone release. Jazani et al. [37] demonstrated that the gut microbiota activates the hypothalamic-pituitary-adrenal (HP2020-10-25A) axis and increases the secretion of serotonin and other neurotransmitters. Onal et al. [38] reported that Bifidobacteriaceae, Lactobacillaceae, and Prevotellaceae species can synthesize neurotransmitters such as γ-aminobutyric acid (GABA) and acetylcholine (ACh) and can promote production of intestinal incretin, glucagon-like peptide-1 and -2 (GLP-1, -2), and the gut hormone peptide YY. GABA is known to stimulate natriuresis and to suppress renal sympathetic nerve activity [9,38]. ACh and GLP-1 have been reported to increase the GFR by renal vasodilation and reducing angiotensin II [7,9,37]. Therefore, gut dysbiosis with reduction of bacteria species such as Bifidobacteriaceae and Lactobacillaceae in CKD causes activation of the RAAS and augmentation of sympathetic outflow and, ultimately, leads to HTN and CKD progression. In addition, changes in GLP and peptide YY can influence energy homeostasis by reduced energy expenditure, lipolysis, and insulin sensitivity and secretion, leading to obesity, hypercholesterolemia, insulin resistance, and/or DM. These are important risk factors for CKD development and can also contribute to CKD progression [4,33,38].
The clinical impact of dysbiosis on CKD-related complications
Increasing evidence supports the concept that dysbiosis in CKD patients is associated with CKD-related complications such as HTN, cardiovascular events, mineral and bone disorder (MBD), and cognitive dysfunction [33,38]. The shift towards a predominantly proteolytic fermentation pattern in CKD promotes the increase of microbiota-derived toxins and increases systemic inflammation.
Cardiovascular disease
Cardiovascular disease is a main cause of morbidity and mortality in CKD patients. Beyond the gut-kidney axis, several reports suggest that diverse mechanisms including enhanced ROS production, leukocyte activation, pro-inflammatory cytokine production, myocyte hypertrophy, and RAAS activation contribute to the development and progression of cardiovascular disease, calling these relationships between the gut and heart the gutheart axis [7,38-41]. Lin et al. [42] showed that elevated levels of pCS and IS are associated with increased mortality in CKD patients while pCS, but not IS, is associated with an increased risk of cardiovascular events. A recent meta-analysis studying TMAO in 19,256 patients also demonstrated that elevated levels of TMAO and its precursors were associated with a 1.7-fold increased risk of major acute cardiovascular events and all-cause mortality in comparison with low TMAO [43].
Cognitive dysfunction
Psychiatric disease is common in CKD patients and is associated with increased morbidity and mortality [9,39,44,45]. Although estimates vary, studies have shown that combined depression and cognitive dysfunction is present in approximately 20% to 25% of CKD patients [7-9,44,45]. Growing evidence suggests that gut dysbiosis promotes dysregulation of the HPA axis with overproduction of glucocorticoids, alterations in levels of neurotransmitters, and activation of pro-inflammatory cytokines, called the gut-brain axis [39,45-48]. Gut microbiota-derived toxins play an important role in the pathogenesis of cognitive dysfunction by direct toxicity or other putative factors such as oxidative stress, inflammation, endothelial dysfunction and vascular calcification [7,45,46]. IS crosses the blood-brain barrier through the organic anion transporter-3 (OAT-3), and may accumulate in the brain in CKD due to dysfunctional OAT-3 [46]. Elevated IS induces inflammation and apoptosis of human astrocytes and glial cells through oxidative stress induction and MAP kinase pathway inhibition [45-48]. Aryl hydrocarbon receptor (AhR), known as a ligand-activated transcriptional factor, is mainly expressed in the hippocampus, cerebral cortex, and cerebellum, and has been implicated in sensorimotor and cognitive functions [7,9,47,48]. Elevated IS activates AhR in astrocytes, which is likely to promote further oxidative stress, leading to sensorimotor and cognitive dysfunction in CKD [47,48]. In a clinical study with 260 hemodialysis (HD) patients, Lin et al. [47] showed that the circulating free form of IS is significantly associated with lower cognitive function test scores, particularly in cognitive abilities screening instrument domains of memory, mental manipulation, and language ability. Further clinical studies based on molecular regulation in the expression of uremic toxins are needed in CKD patients.
CKD-MBD
CKD-MBD is a syndrome recently re-named to embody the biochemical, skeletal, and cardiovascular pathophysiology, not limited to bone disease [49]. CKD-MBD is associated with poor outcomes in terms of myocardial infarction, stroke, bone fracture, and all-cause mortality [50]. Growing evidence indicates elevated uremic toxins are involved in the development of bone abnormalities in CKD [51]. Mozar et al. [50] demonstrated that IS inhibited both osteoclast differentiation and bone-resorbing activity in monocyte/macrophage cellular models. Nii-kono et al. [52] also found that IS suppressed parathyroid hormone (PTH)-stimulated intracellular cyclic adenosine monophosphate production and decreased PTH receptor expression in a primary osteoblast culture from mouse calvaria. In a clinical study with 47 HD patients, Goto et al. [53] showed that IS correlated negatively with alkaline phosphatase, particularly bone-specific alkaline phosphatase, regardless of intact PTH activity. These findings strongly suggested that IS not only inhibits osteoblast function but also has an inhibitory effect on osteoclasts and PTH and, thus, could affect bone remodeling in CKD patients.
TARGETED TREATMENTS IN THE CONTEXT OF GUT MICROBIOTA
Recent evidence about the pathogenic consequences of gut dysbiosis on CKD supports the concept that restoring symbiosis in the gut could be an effective and beneficial targeted treatment for CKD. This would have the additional significance of being more physiological in that it is related to basic diet and digestion. In preclinical or clinical investigations potential therapeutic options have focused on diet modification with healthy supplements, oral adsorbents preventing gut absorption of uremic toxins, modification of the microbiota composition, and modulation of influx/efflux renal transporters for uremic toxins (Table 1). Of these, the treatments currently most commonly used in clinical practice are described below.

Potential therapeutic interventions on the aberrant axis of gut microbiota and chronic kidney disease
High fiber diets
Resistant starch (RS) is a type of carbohydrate that is incompletely digested by human pancreatic amylases. High amylose maize-resistant starch type 2 (HAMRS2), a kind of RS found in starchy foods such as potato, corn, and banana, reaches the large intestine and serves as an energy source for beneficial bacteria such as Bifidobacterium and Lactobacillus [54,55]. Several studies have demonstrated the beneficial effects of HAMRS2 on CKD progression, decreased microbial diversity, and an increased Bacteroidetes-to-Firmicutes ratio in HAMRS2-fed rats [54-56]. Vaziri et al. [56] also reported a favorable effect of this type of RS on reducing oxidative stress and inflammation, and restoring intestinal epithelial tight junctions in a CKD rat model. Krishnamurthy et al. [57] revealed that high fiber diets showed low inflammation and decreased all-cause mortality in a study analyzing data from 14,543 participants in the National Health and Nutrition Examination Survey III. Despite these potential renoprotective effects, high fiber diets have a limitation in alimentotherapy for patients with advanced stages of CKD due to their potassium and phosphorus content. Therefore, practical cooking techniques and counselling should be provided for safe intake in CKD patients.
Prebiotics, probiotics, and synbiotics
Prebiotics are nondigestible food ingredients that can help stimulate the growth of the selected or a limited number of bacteria in the colon [58,59]. Inulin, fructo-oligosaccharides, galacto-oligosaccharides, soya-oligosaccharides, xylo-oligosaccharides, and pyrodextrins are commonly used as prebiotics. They promote the growth of Bifidobacteria and Lactobacilli species, while suppressing other groups of bacteria such as Bacteroides, Clostridia, and Enterobacteria species [59]. Meijers et al. [60] also demonstrated that serum concentrations of IS and pCS were significantly reduced by the oral intake of oligofructose-enriched inulin in 22 HD patients. The prebiotic lactulose also improved kidney function by modifying gut microbiota and inhibiting the production of uremic toxins in 10-week-old adenine-induced CKD Wistar/ST male rats. In addition, lactulose decreased serum levels of creatinine and blood urea nitrogen (BUN) and ameliorated CKD progression by suppressing tubulointerstitial fibrosis [61]. Probiotics are defined as live microorganisms that confer a health benefit on the host when administered in adequate amounts [62]. Probiotics commonly consist of living bacteria such as Bifidobacteria species, Lactobacilli and Streptococci [63]. A pilot clinical trial in patients with CKD stages 3 and 4 showed significantly decreased BUN and improved quality of life after treatment with the Renadyl (Kibow Biotech Inc., Newtown Square, PA, USA) formulation of Lactobacillus acidophilus, Streptococcus thermophilus, and Bifidobacterium longum over 6 months [63]. However, the follow-up randomized controlled trial in 22 patients failed to show decreased uremic toxins or the sensation of enhanced well-being [64]. Some of the benefits of probiotics could be explained by persistent uremia-induced alterations in the gut biochemical milieu as well as by dietary or medicinal regimens that produce an unfavorable microenvironment for the symbiotic microbiota. Synbiotics are the combination regimen of prebiotics and probiotics. A randomized clinical trial by Guida et al. [65] showed a suppressive effect of short-term synbiotic treatment on plasma p-cresol levels without improvement of gastrointestinal symptoms in 30 CKD patients after 4 weeks. A recent multicenter study in 42 HD patients showed an improvement of gastrointestinal symptoms and decreased C-reactive protein after 2 months of treatment [66]. The recent randomized controlled trial of the effects of pre/pro/synbiotics is summarized in Table 2 [63,67-76].
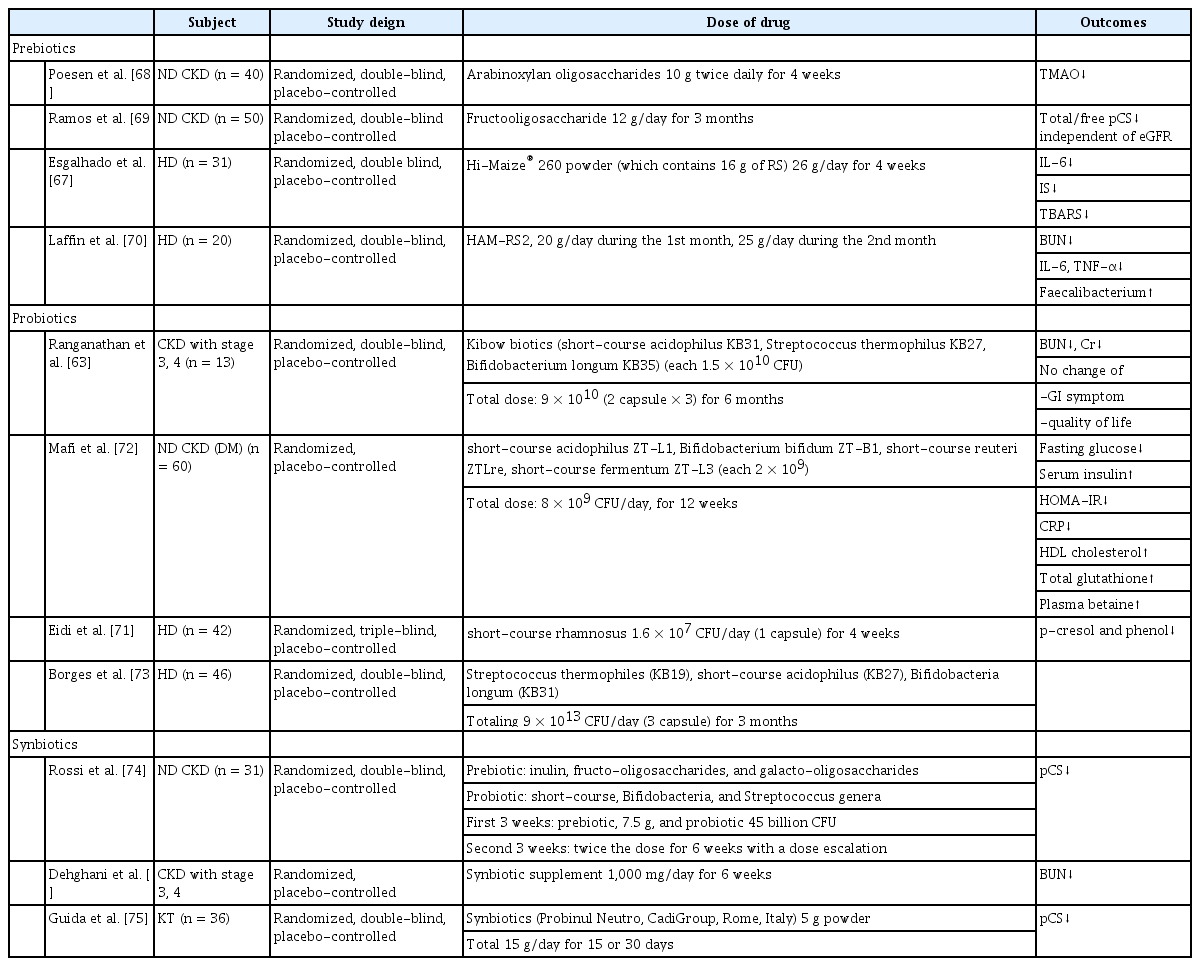
Randomized clinical trials of prebiotics, probiotics and synbiotics in CKD
Fecal microbiota transplantation
Fecal microbiota transplantation (FMT), also known as a stool transplant, is another option to modify the gut microbiota [77,78]. However, there are few data about FMT for the treatment of CKD, except the use of FMT for the treatment of Clostridium difficile infection, which is a common complication in patients with CKD undergoing HD [77,78]. Further research is needed to understand how FMT affects the progression of CKD.
Adsorption therapy
AST-120, an oral charcoal adsorbent, is widely used as a therapeutic agent in CKD patients to absorb circulating uremic toxins and precursors such as IS. Several animal studies have shown that, in addition to the change in composition of the microbiota, AST-120 decreased serum IS levels, reduced the production of ROS by endothelial cells, and impeded subsequent oxidative stress and inflammation [79,80]. Several clinical studies have also shown that AST-120 reduced uremic symptoms, proteinuria and time-to-dialysis in CKD patients [79,81]. However, a recent meta-analysis reported that AST-120 reduced serum IS levels but showed no significant improvement of renal function and all-cause mortality [82]. Sevelamer (hydrochloride or carbonate), a polyphosphate binder, is noted for its ability to bind uremic toxins. Although some experiments and/or clinical studies report beneficial effects on reducing uremic toxins, such as serum IS and pCS levels, the results of the clinical studies published to date are conflicting [83].
CONCLUSIONS
The microenvironmental interface between the gut microbiota and kidney is representative of the relationship that maintains the metabolic and immunologic milieu in humans. Uremic toxins, LPSs as endotoxins, disrupted intestinal barriers, and immune dysregulation are responsible for the bidirectional imbalance in which CKD predisposes to an alteration of gut microbial elements, and the resulting gut dysbiosis induces stage progression and the complications of CKD and, consequently, the effect of being trapped in a reciprocally vicious cycle. Clinical interventions designed to restore the imbalance of gut-kidney symbiosis have emerged as possible treatment options such as acceptable diets including high fiber meals, prebiotic, probiotic, and synbiotic supplementations, adsorbent agents, and FMT. Further clinical studies are required to confirm the safety and efficacy of these novel approaches to the improvement of disease outcomes and survival of patients with CKD.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This study was financially supported by the research fund of Dankook University in 2019.