Upregulation of receptor tyrosine kinase-like orphan receptor 2 in idiopathic pulmonary fibrosis
Article information

Abstract
Background/Aims
Receptor tyrosine kinase-like orphan receptor 2 (ROR2) is a major regulator of Wnt signaling, which is involved in fibroblast dysfunction. Because its role has not been evaluated in idiopathic pulmonary fibrosis (IPF), we examined the clinical implications of ROR2 expression.
Methods
ROR2 mRNA expression was measured using reverse transcription polymerase chain reaction in lung tissue-derived fibroblasts from IPF patients (n = 14) and from controls (n = 10). ROR2 protein was measured using enzyme-linked immunosorbent assay in primary fibroblasts from IPF patients (n = 14) and controls (n = 10), and in bronchoalveolar lavage (BAL) fluids obtained from normal controls (NC; n = 30). IPF patients (n = 84), and other patients with interstitial lung diseases, including nonspecific interstitial pneumonia (NSIP; n = 10), hypersensitivity pneumonitis (HP; n = 10), and sarcoidosis (n = 10).
Results
ROR2 mRNA and protein levels were significantly higher in IPF fibroblasts than in controls (p = 0.003, p = 0.0017, respectively). ROR2 protein levels in BAL fluids from patients with IPF were significantly higher than in those from NC (p < 0.001), and from patients with NSIP (p = 0.006), HP (p = 0.004), or sarcoidosis (p = 0.004). Receiver operating characteristic curves showed a clear difference between IPF and NC in ROR2 protein level (area under the curve, 0.890; confidence interval, 0.829 to 0.950; p < 0.001). ROR2 protein levels were significantly higher in GAP stage III than in GAP stages I and II (p = 0.016).
Conclusions
ROR2 may be related to the development of IPF, and its protein level may be a useful and severity-dependent candidate marker for IPF.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) comprises a group of lung diseases induced by both genetic and epigenetic factors [1]. Among the genetic factors, MUC5B, SFTPC, SFTPA2, RTEL1, TERT, and hTR have been implicated in the development of IPF [2], and global gene expression studies have identified several novel genes expressed in IPF lung tissues [3-6]. Complex interactions among these genes may determine the development and disease progression of IPF [7,8]. Recently, we demonstrated that 178 genes were differentially expressed in IPF lung tissue-derived fibroblasts [9]. Of these, mRNA expression of receptor tyrosine kinase-like orphan receptor 2 (ROR2) was more than 20-fold higher in IPF fibroblasts than in cells from control lungs.
WNT5A is a member of the Wnt family of secreted glycoproteins, which have crucial roles in many cellular activities, including proliferation, differentiation, migration, cell polarity, and cell adhesion [10]. WNT5A is an important regulator of fibroblast proliferation with resistance to apoptosis [11], a characteristic seen in IPF. ROR2 acts as a receptor or co-receptor for WNT5A, and mediates non-canonical Wnt signaling [12]. In the submandibular gland, ROR2-mediated signaling is activated in response to tissue damage, and is associated with the progression of fibrosis [13]. However, its expression has not been evaluated in patients with IPF. Thus, in the present study, we evaluated the relationship between ROR2 expression and clinical characteristics of IPF, assaying ROR2 mRNA and protein levels in fibroblasts and bronchoalveolar lavage (BAL) fluids from normal controls (NC), patients with IPF, and patients with other interstitial lung diseases (ILDs).
METHODS
Study subjects
Primary lung fibroblasts and BAL fluids from patients with diffuse ILDs were obtained from the biobank of Soonchunhyang University Hospital, Bucheon, Korea, after approval of the study protocol by the Ethics Committee of Soonchunhyang University Hospital (IRB No: SCHCA-IRB-2018–10–034 and 201910-BR-058). NCs BAL fluids were obtained from the general population and from hospital personnel, after approval by the hospital Ethics Committee (IRB No: SCHBC 2015–08–025–005). Informed written consent for study participation and sample donation was obtained from each subject. The diagnostic criteria for IPF, hypersensitivity pneumonitis (HP), nonspecific interstitial pneumonia (NSIP), and sarcoidosis were based on international consensus statements [14-22]. The clinical and laboratory data are summarized in the Supplementary material.
Fibroblast culture
Primary lung fibroblasts were cultured from surgical lung specimens of 14 IPF patients and 10 control subjects who underwent surgery for stage I or II lung cancer, as described previously [9]; their clinical characteristics are summarized in Supplementary Table 1 [9,23]. Briefly, lung specimens were finely minced and placed into 150 cm2 cell culture flasks with tissue culture media (TCM) consisting of DMEM (Lonza Inc., Walkersville, MD, USA), 10% fetal bovine serum (Thermo Fisher Scientific Inc., Rockford, IL, USA), 2 mmol/L glutamine, and 1% penicillin-streptomycin-amphotericin (Lonza). Cells were maintained at 37°C in a 5% CO2 incubator and serially subcultured until the fourth passage to yield a morphologically homogeneous population of adherent fibroblasts staining positive for α-smooth muscle actin (SMA; Abcam, Cambridge, MA, USA). Then the cells were stored at −170°C. Fifth-passage fibroblasts (1 × 106) were seeded in 1 mL TCM in 10 cm2 dishes and cultured for 48 hours. After reaching 90% confluence, fibroblasts were washed twice with PBS (Thermo Fisher) and total RNA was extracted using TRI reagent (Ambion, Carlsbad, CA, USA).
Detection of ROR2 mRNA in primary fibroblasts using reverse transcription PCR
Extracted RNA was treated using the Turbo DNA-Free kit (AM1907; Invitrogen, Camarillo, CA, USA). The resulting total RNA (1 μg) was suspended in diethylpyrocarbonate-treated water with 0.5 μg 10 mM dNTPs and oligodeoxythymidine, heated at 65°C for 5 minutes, and then cooled on ice. PCR amplification was performed for 30 cycles (5 minutes at 95°C, 30 seconds at 95°C, 30 seconds at 58°C, and 30 seconds at 72°C) with a final extension at 72°C for 7 minutes. The following primer sequences were used: ROR2, (sense) 5′-TACGCATGGAACTGTGTGAC-3′ and (antisense) 5′-GAGGGGCATTTCCATGTCTTG-3′; β-actin, (sense) 5′-GGACTTCGAGCAAGAGATGG-3′ and (antisense) 5′-AGCACTGTGTTGGCGTACAG-3′. The PCR products were separated on a 1.0% agarose gel containing ethidium bromide in Tris-borate ethylenediaminetetraacetic acid (EDTA) buffer at 100 V for 40 minutes and visualized under ultraviolet light. ROR2 band intensities were normalized to those of β-actin.
Measurement of ROR2 protein in fibroblasts and BAL fluid using enzyme-linked immunosorbent assay
Primary lung fibroblast cells lysed in RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA) with protease-inhibitor (Thermo Fisher) were analyzed using an ROR2 ELISA kit (MyBioSource, San Diego, CA, USA), and protein concentrations were measured using a BCA kit (Thermo Fisher). BAL was collected either from lung segments exhibiting the greatest disease involvement on high-resolution computed tomography (HRCT) in the absence of immunosuppressive therapy, or from the right middle lobe of NC, as described previously [24,25]. Total cell counts were performed using a hemocytometer. Cells were collected by centrifugation (500 × g, 5 minutes), and the supernatants were stored at –80°C. Differential cell counts were performed on 500 cells from BAL fluids, on slides prepared using a cytocentrifuge and Diff-Quik staining, and ROR2 protein levels were measured using an ROR2 ELISA kit according to the manufacturer’s recommendations. The lower limit of detection was 0.25 ng/mL, and values below this limit were set to 0. The inter- and intra-assay coefficients of variation were less than 15%.
GAP stage
Total GAP scores were calculated using the method of Ley et al. [26]. The following four clinical variables were examined to determine the GAP score: sex (0 points for female, 1 point for male), age (0–2 points), forced vital capacity (FVC)% (0–2 points), and diffusing capacity of lung for carbon monoxide (DLCO)% (0–3 points). Patients were divided into eight groups, according to their GAP scores: group 0 (n = 4), group 1 (n = 11), group 2 (n = 20), group 3 (n = 12), group 4 (n = 15), group 5 (n = 11), group 6 (n = 7), and group 7 (n = 4). DLCO was not measured in eight subjects with IPF and thus was excluded from the determinations of GAP scores. GAP scores divided the patients into three stages: GAP stages I (groups 0–3, n = 47), II (groups 4–5, n = 26), and III (groups 6–8, n = 11) (Supplementary Table 2).
Statistical analyses
Data were analyzed using IBM SPSS version 20.0. (IBM Corp., Armonk, NY, USA). The normality of data distribution was tested using the Shapiro–Wilk test. Categorical values were compared using chi-square test and Fisher exact tests. Comparisons were performed using the Kruskal-Wallis test and a post hoc analyses; a Mann-Whitney U test was conducted for comparisons between two groups. Correlations between ROR2 level and other parameters were analyzed using Spearman’s correlation coefficient. Receiver operator characteristic (ROC) analyses were performed to define cut-off values of the scores. The best cut-off values were calculated using the highest Youden’s index. Comparison between different areas under the curve (AUC) was performed using the Z statistic calculation, according to the method of DeLong et al. [27], using MedCalc Statistical Software version 12.2.1.0 (MedCalc Software bvba, Ostend, Belgium). Data are presented as medians with 25% and 75% quartiles for variables with a skewed distribution, or as means ± standard errors of the mean for variables with a normal distribution. Values for p < 0.05 were considered statistically significant.
RESULTS
Clinical characteristics of study groups
Primary fibroblasts were obtained from 14 IPF patients and 10 NC who underwent lung surgery. Clinical information and laboratory data for these subjects have been summarized in a previous study and in Supplementary Table 1 [9]. BAL fluid samples were obtained from patients with IPF (n = 84), NSIP (n = 10), HP (n = 10), and sarcoidosis (n = 10); clinical characteristics of these patients are summarized in Table 1. Patients with IPF had significantly lower FVC and forced expiratory values (FEV1) compared to NC (p < 0.05).
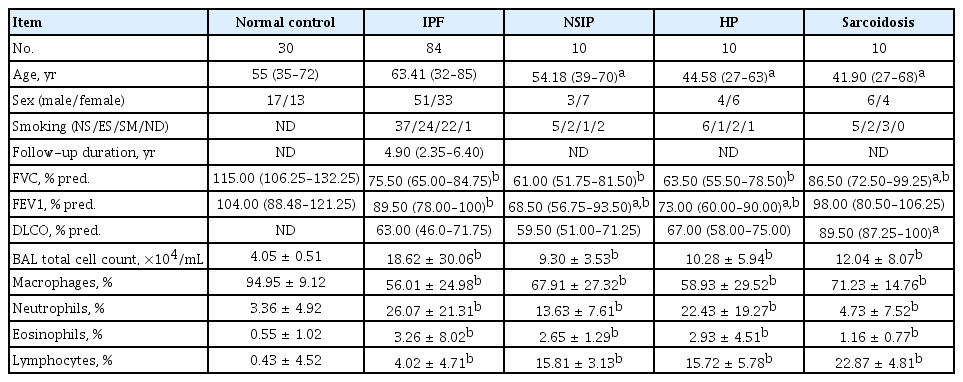
Clinical characteristics of the study subjects
ROR2 mRNA in primary fibroblasts and protein levels in fibroblasts and BAL fluids
The ROR2 mRNA and protein levels in fibroblasts were two-fold higher in the IPF patients than in controls, by reverse transcription PCR and ELISA (p = 0.003 and p = 0.0017, respectively) (Fig. 1). In BAL fluids, there were significant differences in ROR2 protein levels among the NC (0.014 ng/mL [range, 0 to 0.053 ng/mL]), IPF (1.934 ng/mL [range, 0.347 to 9.252 ng/mL]), NSIP (0 ng/mL [range, 0 to 1.866 ng/mL]), HP (0.127 ng/mL [range, 0 to 0.793 ng/mL]), and sarcoidosis (0 ng/mL [0 to 1.866 ng/mL]) groups (Fig. 2A). ROR2 levels were significantly higher in IPF patients than in NC (p < 0.001), HP patients (p = 0.004), NSIP patients (p = 0.006), and sarcoidosis patients (p = 0.004). The ROC curve showed a clear difference between IPF patients and NC (AUC, 0.890; 95% confidence interval [CI], 0.82 to 0.950) (Fig. 2B) and between IPF patients and those with other ILDs (n = 30; AUC, 0.718; 95% CI, 0.660 to 0.866) (Fig. 2C). A cut-off level of 0.305 ng/mL gave 93.3% specificity and 76.2% sensitivity for distinguishing between NC and IPF patients. A cut-off level of 0.082 ng/mL exhibited 53.3% specificity and 88.1% sensitivity for distinguishing between IPF and other ILDs. There were no differences in plasma ROR2 levels between 37 NC and 90 IPF patients (p = 0.105) (Supplementary ig. 1A), and no correlation between ROR2 concentrations in plasma and those in BAL fluids of 60 IPF patients (p = 0.923) (Supplementary Fig. 1B).
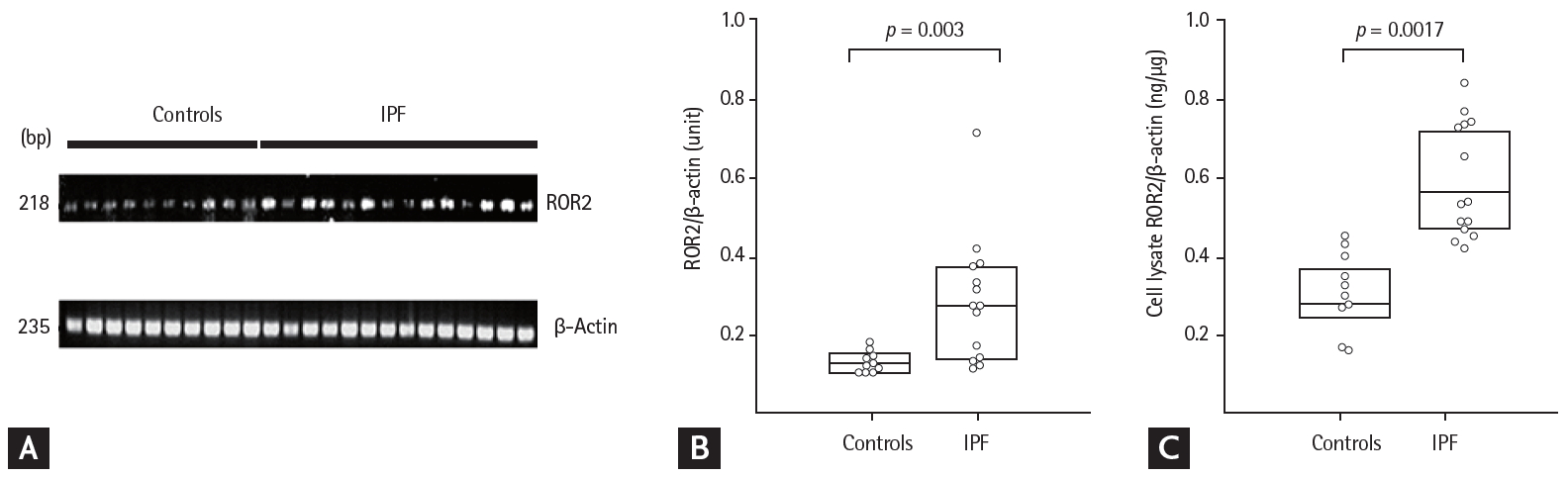
Receptor tyrosine kinase-like orphan receptor 2 (ROR2) mRNA and protein levels in lung fibroblasts from 14 idiopathic pulmonary fibrosis (IPF) patients and 10 controls. (A) Reverse transcription PCR and (B) densitometric analyses of ROR2 band intensities after normalization to those of β-actin. (C) ROR2 protein in cell lysates. The data are presented as medians with 25% and 75% quartiles.
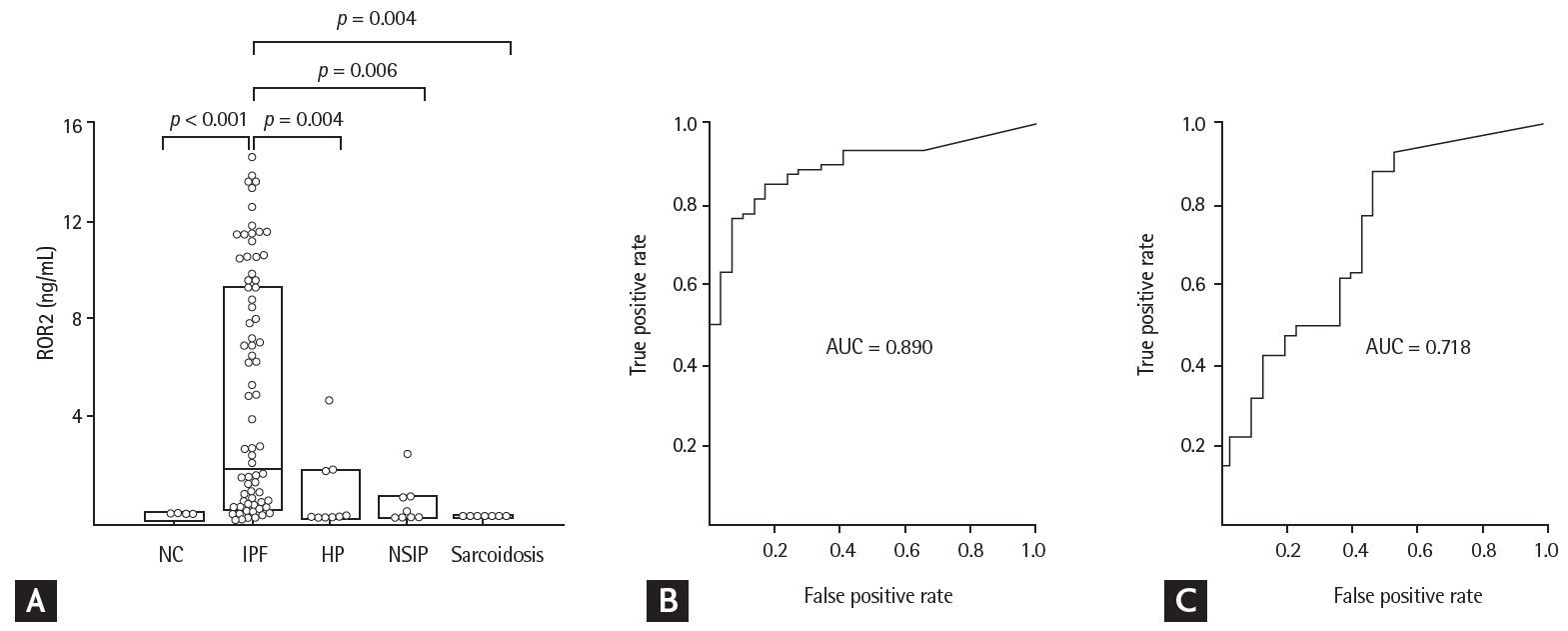
Receptor tyrosine kinase-like orphan receptor 2 (ROR2) protein concentrations in bronchoalveolar lavage (BAL) fluids and receiver operating characteristic (ROC) analyses among study groups. (A) ROR2 protein was detected in 15 of 30 normal controls (NC), 78 of 84 idiopathic pulmonary fibrosis (IPF) patients, 4 of 10 hypersensitivity pneumonitis (HP) patients, 4 of 10 nonspecific interstitial pneumonia (NSIP) patients, and 0 of 10 sarcoidosis patients. The data are presented as median values with 25% and 75% quartiles. (B) ROC curve of ROR2 protein levels in IPF patients and NC. A cut-off value of 0.305 ng/mL had an area under the curve (AUC) of 0.890, a specificity of 93.3%, and a sensitivity of 76.2% for differentiating IPF patients from NC. (C) ROC curve of the ROR2 protein level in patients with IPF and those with other interstitial lung diseases (ILDs). A cut-off value of 0.082 ng/mL had an AUC of 0.763, a specificity of 53.3%, and a sensitivity of 88.1% for differentiating IPF patients from those with other ILDs.
ROR2 protein levels and clinical characteristics
Patients were divided into eight groups according to their GAP score: group 0 (n = 4), group 1 (n = 11), group 2 (n = 20), group 3 (n = 12), group 4 (n = 15), group 5 (n = 11), group 6 (n = 7), and group 7 (n = 4) (Supplementary Table 1). There were significant differences in ROR2 protein levels by GAP stage (stage I vs. II vs. III: 1.718 ng/mL [range, 0.193 to 8.613 ng/mL] vs. 1.015 ng/mL [range, 0.322 to 4.674 ng/mL] vs. 10.547 ng/mL [range, 3.989 to 11.593 ng/mL]; p = 0.016) (Fig. 3A and Supplementary Table 3). There were no differences in ROR2 protein levels with smoking status (NS vs. ES vs. SM: 1.627 ng/mL [range, 0.264 to 7.034 ng/mL] vs. 1.593 ng/mL [range, 0.379 to 8.954 ng/mL] vs. 3.347 ng/mL [range, 0.788 to 9.457 ng/mL]) (Fig. 3B), or sex (males vs. females: 1.718 ng/mL [range, 0.45 to 9.544 ng/mL] vs. 2.762 ng/mL [range, 0.24 to 8.006 ng/mL]) (Fig. 3C). ROR2 protein level was not correlated with differential cell counts in BAL fluid, initial FVC and DLCO, or changes in those parameters during follow-up (Supplementary Table 4).
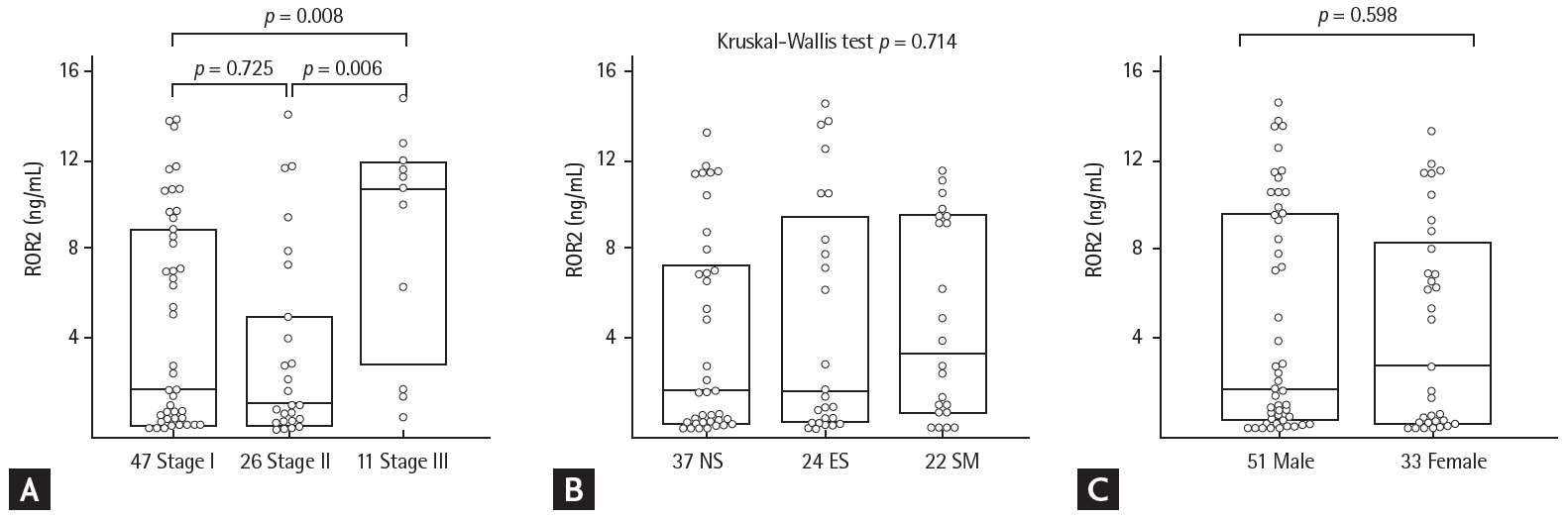
Comparisons of receptor tyrosine kinase-like orphan receptor 2 (ROR2) protein concentrations in bronchoalveolar lavage fluids from idiopathic pulmonary fibrosis patients according to (A) GAP stage, (B) smoking status, and (C) sex. NS, never-smoker; ES, ex-smoker; SM, smoker.
Correlation analyses between ROR2 and genes associated with Wnt signaling
Correlation analyses were performed on expression of the ROR2 gene with genes related to the Wnt signaling pathway in IPF fibroblasts (p < 0.05) (Fig. 4 and Supplementary Table 5). Among the 15,020 genes expressed by fibroblasts [9], 22 genes associated with Wnt pathways were significantly correlated with ROR2 gene expression. This finding indicates that the ROR2 gene may have a synergistic regulatory effect on lung fibrosis by acting with other Wnt signaling genes.
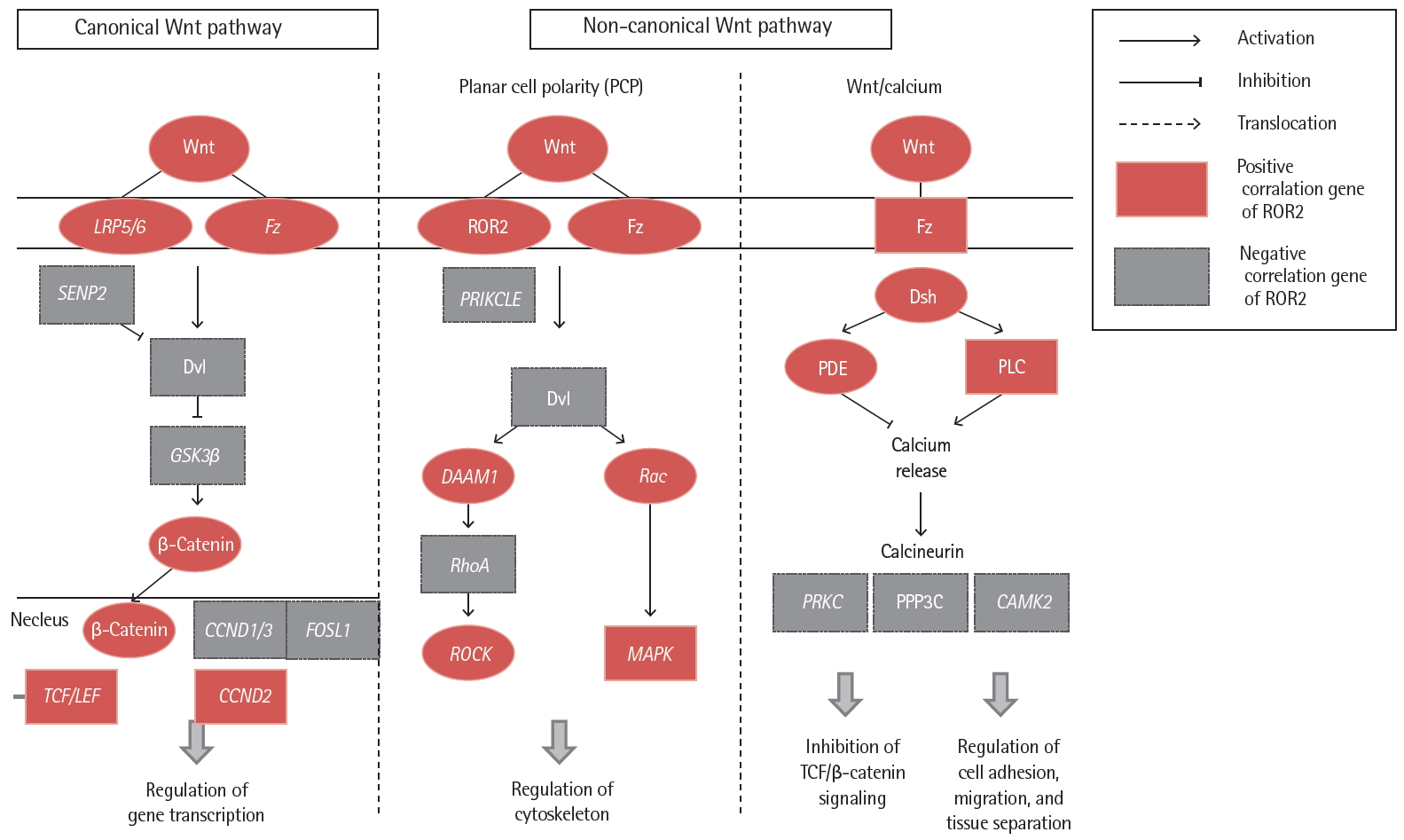
Correlation of idiopathic pulmonary fibrosis fibroblast gene expression between receptor tyrosine kinase-like orphan receptor 2 (ROR2) and Wnt signaling pathway genes. LRP5/6, low density lipoprotein receptor-related protein 5/6; Fz, frizzled; SENP2, sentrin-specific protease 2; Dvl, dishevelled segment polarity protein; GSK3β, glycogen synthase kinase 3 beta; CCND, cyclin D1; FOSL1, FOS like 1, AP-1 transcription factor subunit; TCF, T-cell factor; LEF, lymphoid enhancer factor; PRIKCLE, prickle planar cell polarity protein; DAAM1, disheveled-associated activator of morphogenesis 1; RhoA, Ras homolog family member A; ROCK, Rho associated coiled-coil containing protein kinase; Rac, Rac family small GTPase 1; MAPK, mitogen-activated protein kinase; Dsh, dishevelled; PDE, phosphodiesterase; PLC, phospholipase C gamma; PRKC, serine/threonine-protein kinase PrkC; PPP3C, protein phosphatase 3 catalytic subunit; CAMK2, Ca2+/calmodulin-dependent protein kinase II.
DISCUSSION
We demonstrated that ROR2 gene and protein expression were significantly increased in the primary fibroblasts of IPF patients compared to controls, and that ROR2 protein in IPF BAL fluids was also significantly elevated, relative both to controls and to patients with other ILDs. In addition, IPF was differentially diagnosed from NC and from other ILDs at ROC cut-off levels for ROR2 of 0.305 ng/mL and 0.082 ng/mL, respectively. Clinically, ROR2 protein levels were significantly increased in more severe manifestations of IPF, including GAP stage III. To the best of our knowledge, this is the first evidence suggesting that ROR2 expression is related to the development and severity of IPF and that ROR2 protein level may be a surrogate biomarker for differentially diagnosing IPF from other ILDs. MMP-1, MMP-7 [28], CCL8 [9], CCL18 [29], YKL-40 [30], SPA, SP-D [31], and KL-6 [32] have been proposed as biomarkers of IPF. In a previous study, plasma MMP-1 and MMP-7 levels were significantly higher in IPF patients than in those with other lung diseases, such as chronic obstructive pulmonary disease and sarcoidosis, with a sensitivity of 96.3% and a specificity of 87.2% for the differential diagnosis of IPF from HP [28]. In a previous study, we reported 178 differentially expressed genes in fibroblasts derived from IPF lungs [9]. Among the top 15 genes, ROR2 expression was >20-fold higher in IPF fibroblasts. In that study, we performed analyses using GEO datasets (Sridhr’s study using primary fibroblasts [n = 1,813, 4 controls and 10 IPF subjects; GSE44723] and Ronzani’s study [n = 3, 5 controls and 5 IPF subjects; GES45686]). Compared to the 178 genes identified in our study, only 9 genes (MOXD1, PITX1, POSTN, TMEM51, CDC42EP3, UBE2K, ALDH3A2, NBEAL2, and IGFBP2) overlapped, and ROR2 was not among them. This may be due to biological differences between the primary fibroblasts used in our study and uncultured cells isolated from IPF lungs.
ROR2 is a transmembrane protein with normal expression in the developing heart, brain, and lungs [33,34]. It is localized mainly near the plasma membrane [35], and was easily detected in BAL fluids in our study. In addition, ROR2 level was not correlated with the cellular profile of BAL fluid or with lung function profile or age. In post hoc analyses, there were no significant differences in ROR2 levels with sex or smoking status (Fig. 3). The potential role of ROR2 in Wnt signaling has recently been revealed. Wnt signaling is mediated through both beta-catenin-dependent and independent pathways [36]. Multiple receptors, including Frizzled (Fzd), Lrps, ROR2, and Ryk, are involved in Wnt signaling. Wnt ligands interact with the Fzd family of seven-span transmembrane receptor molecules and co-receptors of the low-density lipoprotein receptor-related protein (Lrp) family [37]. The cysteine-rich domains (CRDs) of ROR2 interact with Wnt ligands and the CRDs of Fzd2 in the N-terminal extracellular region [38]. ROR2 acts as a receptor or co-receptor for WNT5A, and mediates non-canonical Wnt signaling, including the inhibitory effects of WNT5A on WNT3A-mediated activation of the canonical Wnt pathway [12,39]. WNT5A is a key regulator of epithelial-mesenchymal interactions during lung development and maturation [40], and is an important regulator of fibroblast proliferation with resistance to apoptosis [11,41], both of which are characteristic of IPF. WNT5A signaling is also involved in producing pro-inflammatory cytokines, such as interleukin (IL)-12 and IL-6 [42,43]. Thus, increased levels of ROR2 may modulate Wnt/β-catenin signaling, leading to increased numbers of fibroblasts via inhibition of apoptosis.
One limitation of our study was the small number of subjects, particularly those with other ILDs, including NSIP, HP, and sarcoidosis. In addition, long-term follow-up will be necessary to fully address the clinical implications of ROR2, particularly the survival rate of IPF. Furthermore, validation studies using other cohorts are required to improve the diagnostic utility of ROR2.
In conclusions, ROR2 mRNA and protein levels were significantly higher in IPF fibroblasts, and ROR2 protein was significantly elevated in the BAL fluid of patients with IPF, compared to NC and those with other ILDs, including HP, NSIP, and sarcoidosis. Cut-off ROR2 levels of 0.305 and 0.082 ng/mL offered good accuracy with high specificity and sensitivity for differentiating IPF from NC and from other ILDs. Our data suggest that ROR2 has potential for use as a surrogate diagnostic marker.
KEY MESSAGE
1. Receptor tyrosine kinase-like orphan receptor 2 (ROR2) level was significantly higher in bronchoalveolar lavage f luid of patients with idiopathic pulmonary fibrosis (IPF).
2. ROR2 offered good diagnostic accuracy with high specificity and sensitivity for differentiating IPF from normal control.
3. ROR2 levels were significantly higher in GAP stage III than in GAP stages I and II.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2020R1I1A1A01067088). And a Soonchunhynag University grant to Jong Sook Park. The samples were generously provided by Soonchunhyang University, Bucheon Hospital Biobank, a member of the National Biobank of Korea, supported by the Ministry of Health, Welfare, and Family Affairs, Republic of Korea.
Supplementary Material
Supplementary Table 1.
Clinical characteristics of the study subjects providing fibroblasts for culture
Supplemental Table 4.
Correlation of ROR2 levels with BAL fluid cell and lung function profiles in IPF
Supplemental Table 5.
Correlation of ROR2 protein concentration with WNT pathway-related gene expression in IPF patients (p < 0.05)
Supplementary Figure 1.
Receptor tyrosine kinase-like orphan receptor 2 (ROR2) concentrations in plasma and its correlation with ROR2 levels in bronchoalveolar lavage fluids (BALFs). (A) ROR2 concentrations in plasma from normal controls (n = 37) and idiopathic pulmonary fibrosis (IPF) subjects (n = 90). (B) Correlation of paired samples between plasma and BALFs from IPF subjects (n = 49). The data are presented as median values with 25% and 75% quartiles.