Recent advances in the diagnosis and management of interstitial pneumonia with autoimmune features: the perspective of rheumatologists
Article information

Abstract
Interstitial pneumonia with autoimmune feature (IPAF) is a recently established disease entity that is comprised of interstitial lung diseases with evidence of autoimmune features but that does not fulfill the criteria for definite autoimmune rheumatic diseases. The classification criteria for IPAF were defined by the European Respiratory Society and American Thoracic Society in 2015. However, further studies to establish IPAF subgroups and treatment modalities for each subgroup are still needed. In this review, we discuss recent advances regarding IPAF and raise critical points for the diagnosis and management of patients with IPAF from the perspective of rheumatologists.
INTRODUCTION
Interstitial lung disease (ILD) is a group of lung diseases that affect the interstitium with inflammatory and fibrotic insults. ILD is divided into four different categories according to etiology as follows: (1) occupational/environmental factors; (2) iatrogenic factors; (3) factors associated with connective tissue diseases (CTDs; as in autoimmune rheumatic disease [ARD]); and (4) idiopathic factors [1]. The concept of ILD with undifferentiated CTD was first proposed by Kinder et al. [2]. Thereafter, the European Respiratory Society (ERS) and American Thoracic Society (ATS) Task Force on Undifferentiated Forms of CTD-associated ILD designated this disease as interstitial pneumonia with autoimmune feature (IPAF) and established the classification criteria for IPAF [3]. A certain portion of previously diagnosed idiopathic ILD cases can now be reclassified as IPAF. However, the current classification criteria for IPAF have several limitations as they do not include critical clinical domain items and the heterogeneity of IPAF cases has been ignored. Here, we review recent advances in the diagnosis and management of IPAF and propose critical points regarding IPAF from the perspective of rheumatologists.
DEFINITION AND CLASSIFICATION CRITERIA FOR IPAF
Cases of currently defined IPAF were formerly classified as idiopathic ILD. Among idiopathic ILDs, some cases show autoimmune traces, which have been referred to, using various terms, as follows: “undifferentiated CTD-associated ILD,” “lung-dominant CTD,” or “autoimmune-featured ILD” [2,4–6]. The former nomenclatures have similarities with regard to distinguishing ILD with autoimmune traces; however, the detailed classification criteria differ from each other. The ERS/ATS Task Force redefined and unified the former nomenclature and newly established classification criteria for IPAF [3]. The fundamental entry criteria for IPAF are as follows: (1) presence of interstitial pneumonia; (2) exclusion of other possible etiologies of ILD; and (3) not fulfilling the definite criteria for ARD [3]. Furthermore, cases that satisfy the entry criteria should fulfill at least two of the following domains of the IPAF classification criteria: (1) clinical domain; (2) serological domain; and (3) morphological domain (Table 1). The usual interstitial pneumonia (UIP) pattern does not satisfy the morphological domain of the classification criteria; however, if the UIP pattern fulfills the other two domains (clinical and serological domains), it can be classified as IPAF. The classification criteria for IPAF require a multidisciplinary approach that involves pulmonologists, rheumatologists, radiologists, laboratory physicians, and pathologists.

Classification criteria of interstitial pneumonia with autoimmune features defined by ERS/ATS
EPIDEMIOLOGY AND PREVALENCE OF THE CLINICAL, SEROLOGICAL, AND MORPHOLOGICAL FINDINGS OF IPAF
The prevalence of IPAF ranges from 7.1% to 34.1% of all ILDs [7–10], and the prevalence is varied because all studies reported to date have been conducted retrospectively. The average age of patients with IPAF is in the mid-60s, and most studies have shown a female predominance [9–15], whereas some have suggested an equal sex distribution [7,8]. IPAF showed a clear difference from idiopathic pulmonary fibrosis (IPF), which shows male predominance. The reported prevalence of former or current smoking history in patients with IPAF ranges from 38.8% to 56.2% [7,11,16,17]. The most common morphological pattern is non-specific interstitial pneumonia (NSIP) in IPAF, with a prevalence of 42.1% to 68.9% [7,8,10,11,13–15,17,18]. However, some reports showed UIP dominance in IPAF, which was found in 27.9% to 44.2% of cases [12,16,19,20].
Among the three domains of the IPAF classification criteria, the morphological domain is the most frequently fulfilled domain (78.9% to 100%), followed by the serological and clinical domains accounting for 48.9% to 93.0% and 31.5% to 62.2% of cases, respectively [7–10,15,17,21]. The most common findings in the clinical domain are Raynaud’s phenomenon (8.89% to 74.1%), mechanic’s hands (4% to 29%), inflammatory arthritis/morning stiffness (16% to 76.5%), and Gottron’s sign (5% to 18%) [7–10,15–17,19]. In the serological domain, antinuclear antibody (ANA) titers ≥ 1:320 in diffuse, speckled, homogeneous patterns or any titer with a nucleolar or centromere pattern are the most frequent findings (10.3% to 82.4%), followed by rheumatoid factor (4.4% to 28.6%), anti-Ro (4.4% to 55.9%), and anti-tRNA synthetase positivity (0% to 35.7%) [7–10,15–17,19].
COMPARISON BETWEEN IPAF AND ARD-ILD
ILD is a relatively common pulmonary manifestation in various ARDs, such as systemic sclerosis (SSc), inflammatory myositis, primary Sjögren’s syndrome (pSS), and rheumatoid arthritis (RA). The highest incidence of ILD in ARDs is found in patients with SSc (43.0% to 91.0%) [22–24], followed by those with inflammatory myositis (19.9% to 78%) [25–27], RA (7.7% to 33.0%) [28,29], and pSS (8.0% to 39.1%) [30–32]. The positive findings of the clinical and serological domains of the IPAF groups demonstrate that the features associated with SSc, RA, inflammatory myositis, and pSS are frequently found in patients with IPAF. Although most features of the clinical and serological domains show similar prevalence rates with ARD-associated ILD (ARD-ILD, formerly CTD-ILD), the SSc-specific autoantibody, anti-topoisomerase (Scl-70), is not predominantly present in the IPAF group (0% to 5.7%) [7–10,15,17,19]. However, in the serological domain, ANA positivity in the IPAF classification criteria includes any titer with a nucleolar or centromere pattern, which is the typical pattern of limited cutaneous SSc [33]. In patients with SSc, anti-topoisomerase antibody positivity is more closely associated with the presentation of ILD [34,35], and this characteristic differs from that observed in the IPAF cohort. This discordance may be due to the difference between IPAF and ARD-ILD.
PATHOPHYSIOLOGY AND GENETIC BACKGROUND OF IPAF
The pathophysiology of IPAF has not been fully elucidated. As included in the nomenclature for IPAF, patients with IPAF are assumed to have features of both idiopathic interstitial pneumonia (IIP) and ARD-ILD, which are represented by idiopathic fibrosis and the autoimmune-mediated inflammatory process (Fig. 1). Several genetic polymorphisms are known to be involved in the pathogenesis of ILD [36,37]. IPF-associated gene mutations are found in patients with telomerase-, surfactant protein-, immune function-, and mucin 5B (MUC5B)-related conditions [37]. Newton et al. [38] demonstrated that the leukocyte telomere length is longer in patients with IPAF and ARD-ILD than in those with IPF. The leukocyte telomere length is associated with pulmonary function and lung transplant outcome in patients with IPAF [38]. Furthermore, MUC5B polymorphism is more common in patients with IPAF than in those with IPF, but the same is not true for Toll-interacting protein (TOLLIP) polymorphism [38]. These findings confirm that IPAF and IPF have similar genetic backgrounds in terms of pathogenesis but show different detailed genetic mutations. Numerous genetic polymorphisms are found in ARDs, such as RA, pSS, and SSc, but there have been no studies regarding the association between ARD-related genes and the development of IPAF. Therefore, a genome-wide association study in patients with IPAF could provide new insights into the pathogenesis of IPAF.

Schematic spectra of interstitial pneumonia with autoimmune feature (IPAF), idiopathic interstitial pneumonia (IIP), and autoimmune rheumatic disease-associated interstitial lung disease (ARD-ILD) and the representative features of IIP and ARD-ILD.
Another issue is the influence of environmental factors on the pathophysiology of IPAF. As mentioned in the previous section, history of smoking is associated with the development of IPAF. In the field of rheumatology, smoking has been shown to play a role in disease progression and even in autoantibody formation. Anti-citrullinated protein antibody positivity is associated with history of smoking in RA [39,40], and former smokers have higher odds ratios for anti-synthetase antibody positivity in inflammatory myositis [41]. Among patients with pSS, those with a history of smoking (current and former smokers) had a higher ANA positivity rate than never smoker [42]. In an animal model of RA, smoking was shown to enhance peptide citrullination in lung tissue and tracheal cartilage [43]. This suggested that smoking can locally promote anti-citrullinated peptide antibody production, especially in the lungs. Although smoking is associated with the development of ILD, it could affect the development of IPAF via autoantibody formation.
SUBCLASSIFICATION OF IPAF
In the classification criteria for IPAF, the items that constitute the clinical and serological domains are derived from the features of specific ARDs. Therefore, it is possible to divide IPAF into several subtypes according to the items of the clinical and serological domains (Table 2). Subgrouping IPAF according to the following four categories, (1) SSc type, (2) arthritis type, (3) myositis type, and (4) systemic lupus erythematosus (SLE)/pSS type, may be considered in future revision of the classification criteria for IPAF. We suggest these IPAF subgroups in the present review because although ARDs have definite similarity with regard to autoimmune-mediated processes, they vary in their detailed pathogenesis, clinical features, and serological markers. Furthermore, subgrouping IPAF could provide insights and concepts for establishing individualized treatment strategies. The 2015 classification criteria include various patients with IPAF but have significant limitations in that they ignore the specific features of each ARD and classify too many heterogenic patients into a single IPAF category.
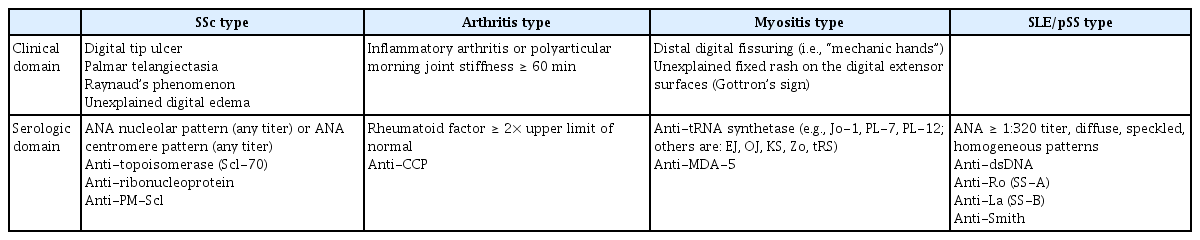
Suggested subclassification of interstitial pneumonia with autoimmune features
POTENTIAL NOVEL DIAGNOSTIC TOOL FOR IPAF
The 2015 ERS/ATS classification criteria for IPAF are significant as they attempt to establish IPAF as a novel disease entity and create a basis for conducting future research on IPAF. However, updating the clinical domains with more objective signs could improve the value of the classification criteria. The inclusion of inflammatory arthritis may increase the validity of the criteria using imaging studies, such as ultrasonography, and limit the involvement of small joints, which are mainly involved in RA [44]. In the clinical domain associated with SSc, adding abnormal nailfold capillaroscopic findings, which are among the classification criteria for SSc, could be considered [45]. Two research studies reported abnormal nailfold capillaroscopic findings in approximately 20% of patients with IPAF [8,10]. Tirelli et al. [18] suggested that a multidisciplinary approach including nailfold capillaroscopy could improve the differentiation between IPAF, ARD-ILD, and idiopathic ILD. Several parameters can be observed in nailfold capillaroscopy for patients with SSc, including (1) irregularly enlarged capillaries, (2) giant capillaries, (3) hemorrhage, (4) loss of capillaries (avascular area), (5) disorganization of the vascular array, and (6) capillary ramifications [46]. In our clinic, we performed nailfold capillaroscopy examinations in patients with IPAF and found abnormalities (Fig. 2). Although the prevalence of ILD is relatively high in pSS, the 2015 classification criteria for IPAF do not include clinical domains related to pSS. The revised 2016 classification criteria for pSS include objective signs of dry eyes and mouth by Shirmer’s test and unstimulated whole saliva flow rate [47]. These are noninvasive and convenient means of checking objective signs of sicca symptoms. The recently revised classification criteria for SLE were updated with a scoring system [48]. High scores are assigned to items on cutaneous lupus symptoms, serositis (pleural and pericardial effusions and pericarditis), and proteinuria [48]. These items could be evaluated by inspection (cutaneous lupus), radiography (serositis), and quantitative urine analysis (proteinuria). In the myositis-associated clinical domains, the most typical clinical presentation and signs of inflammatory myositis are proximal muscle weakness and elevated levels of muscle enzymes (creatinine kinase, lactate dehydrogenase, aldolase, aspartate aminotransferase, and alanine aminotransferase) [49]. Therefore, the typical proximal muscle weakness and elevated levels of muscle enzymes should be considered potential clinical domains of the classification criteria for IPAF in future. Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is a systemic vasculitis mainly involving the small vasculature, and myeloperoxidase (MPO)-ANCA and proteinase 3 (PR3)-ANCA are the most common autoantibodies found in AAV. The prevalence of ILD ranged from 46% to 71% among patients with MPO-ANCA-positive AAV and from 0% to 29% in those with PR3-ANCA-positive AAV [50]. In future, the revised classification criteria for IPAF may be updated by including clinical and serological domains to enhance the usefulness of the criteria by considering the objective signs and examinations used in the recent classification criteria for ARDs.

Abnormal nailfold capillaroscopy findings in a 52-year-old female patient with interstitial pneumonia with autoimmune features with a rheumatoid factor level of 45 IU/mL (reference range, 0 to 18) and an non-specific interstitial pneumonia-dominant morphological pattern. (A) Giant capillary (black arrowhead). (B) Avascular area. (C) Hemorrhage (black arrows).
TREATMENT OF IPAF
The management of IPAF is based on clinical trials of ARD-ILD and IIP due to the lack of controlled interventional clinical trials in patients with IPAF. Usually, immunosuppressants, including systemic glucocorticoid and additional antifibrotic agents, are used for patients with IPAF. The fundamental hypothesis for the use of immunosuppressants in ARD-ILD is that immune and inflammatory reactions are the causes of ILD progression in patients with ARD. Furthermore, alveolar epithelial injury, which is the main pathological process in IIP, is also assumed to be a critical factor in the development of ARD-ILD [51]. Initial glucocorticoid therapy with the addition of glucocorticoid-sparing agents, such as mycophenolate mofetil, azathioprine, cyclophosphamide, and calcineurin inhibitors, is the main treatment modality for ARD-ILD [52]. However, the use of glucocorticoids and immunosuppressants did not significantly reduce the hazard ratio (HR) of mortality in IPAF [7,8,12,14], but Li et al. [53] reported that patients with IPAF who received immunosuppressant therapy showed improvement of the diffusion capacity of the lungs for carbon monoxide (DLCO) based on 6-month follow-up data.
Recently, the antifibrotic agent pirfenidone has been shown to be effective in improving the pulmonary function and survival of patients with IPF [54–56]. Another antifibrotic agent, nintedanib, is a tyrosine kinase inhibitor initially developed as an antitumor agent that suppresses the vascular endothelial growth factor pathway. Nintedanib significantly reduces the decline in forced vital capacity (FVC) and mortality rate in patients with IPF [57,58]. Pirfenidone therapy for SSc-ILD failed to show any beneficial effects on FVC and other clinical parameters (dyspnea score, 6-minute walking distance, and modified Rodnan skin score) [59]. In contrast, nintedanib use in SSc-ILD showed better prognosis than placebo in terms of decline in FVC [60]. In studies on IPAF, antifibrotic agents did not show any beneficial effects in reducing mortality [8,9]. The most important limitation in establishing a treatment strategy for IPAF is that no randomized clinical trials of immunosuppressant and antifibrotic agents for IPAF have yet been conducted. A number of clinical trials have been designed to confirm the therapeutic effects of antifibrotic agents in unclassified ILD, including IPAF [61,62], and these may provide more evidence to support the use of antifibrotic agents in IPAF. The medications used in IPAF patients in previous studies are summarized in Table 3.

Medications used in patients with interstitial pneumonia with autoimmune features
PROGRESSION OF IPAF TO ARD-ILD
The progression of IPAF to definite ARD is an interesting issue. As shown in Fig. 1, IPAF is assumed to have the characteristics of both IIP and ARD-ILD. Four studies reported the progression rates of IPAF to definite ARD and showed that 12.2%, 13.5%, 16%, and 18.8% of IPAF cases had progressed to ARD at 4.5, 2.6, 1, and 5.2 years of follow-up, respectively [11,16,21,63]. Combining the results of the four studies, we found RA in 11 cases, SSc in six, pSS in five, polymyositis in five, AAV in two, overlap syndrome in three, and SLE in one [11,16,21,63]. In the field of rheumatology, the concept of “preclinical ARD” has recently been settled, and close follow-up with early treatment has been discussed [64,65]. In one study, 6 months of treatment with immunosuppressants improved the DLCO significantly in patients with IPAF, supporting the usefulness of early treatment in IPAF [53]. In one study, 14% of RA patients showed ILD prior to arthritis symptoms [66]. Furthermore, ARD patients presenting ILD as the first manifestation can be classified as IPAF. Therefore, assuming IPAF to be a preclinical ARD and starting early treatment should be considered in patients with IPAF.
PROGNOSIS OF PATIENTS WITH IPAF
The prognosis of patients with ILD focuses on the decline of pulmonary function, acute exacerbation (AE) events, and survival rate [67–69]. Pulmonary functions assessed using the DLCO and total lung capacity progress more slowly in patients with IPAF than in those with IIP (−1.21% vs. −4.58% and −0.75% vs. −2.32% predicted/year, respectively), and these are similar when compared between patients with IPAF and ARD-ILD [12]. Collins et al. [20] demonstrated that the DLCO had improved in patients with IPAF but declined in those with IPF or ARD-ILD, and FVC had declined in all the patients at 1-year follow-up. Another study comparing pulmonary function changes over time solely in patients with IPAF showed a continuous decline in FVC from 82% to 70.5% at a median follow-up of 45 months [16]. Chartrand et al. [13] reported that adjusted FVC and DLCO by age, sex, and history of smoking were stable at 4-year follow-up in patients with IPAF. With regard to pulmonary function, patients with IPAF tend to show worsening of FVC but relatively conserved DLCO over time.
Yoshimura et al. [17] reported that patients with IPAF have a higher prevalence of AE events than those with IIP. However, in subgroup analysis comparing AE events between IPAF with a UIP pattern and IIP with IPF, and between IPAF with an NSIP pattern and IIP with NSIP, showed non-significant results [17]. AE events are the leading cause of death in ILD. Therefore, comparison of AE events between IPAF and non-IPAF groups is crucial in predicting survival [69]. In one study, AE events were observed less frequently in patients with IPAF than in those with IPF (25.9% vs. 35.4%, respectively) [9]. However, there have been only a few studies regarding the incidence of AE events in patients with IPAF, and the prevalence rates of AE events in patients with IPAF and IPF remain unclear.
The median survival period is longer in patients with IPAF and ARD-ILD than in those with IPF [9,12,16], and the 5-year survival rate was higher in patients with IPAF than in those with IPF (69.5% vs. 36.8%, respectively, p < 0.001) [16]. There is no significant difference in survival rate between patients with IPAF with a UIP pattern and those with IPF [7,9,17,70]. In one study, patients with IPAF showed better survival than did those with ARD-ILD, including SSc, inflammatory myositis, and RA [13]. Another study demonstrated that patients with ARD-ILD had better survival than those with IPAF [7]. Cox regression analysis showed discrepancies in the impacts of IPAF on mortality. Lim et al. [9] and Yoshimura et al. [17] reported lower HR for the diagnosis of IPAF, whereas Chartrand et al. [13] reported that the form of ILD (IPAF or ARD-ILD) is not a significant predictor of mortality.
The predictive factors of mortality in IPAF are older age, UIP pattern, lower baseline FVC/DLCO, history of smoking, and presence of AE events [7–9,11–14,16,17]. Several studies included the morphological form of ILD as UIP compared with non-UIP, or NSIP compared with the NSIP + organizing pneumonia (OP)/OP pattern. In subgroup analyses, some studies showed that patients with IPAF with a UIP pattern had poorer survival than those with IPAF with a non-UIP pattern [12,70]. However, another study demonstrated no significant difference in overall survival between patients with IPAF with a UIP pattern and those with IPAF with an NSIP pattern [8]. The UIP pattern showed an increased HR in one study (HR, 3.847; 95% confidence interval [CI], 1.991 to 7.434) [12], but other studies showed that the UIP pattern is not a significant factor predicting mortality (HR, 1.53; 95% CI, 0.54 to 4.31) [8,14,16]. Oldham et al. [7] reported that the UIP pattern was associated with increased HR for mortality in univariate analysis but that it lost its significance in multivariate analysis. Other studies divided IPAF cases according to the morphological domain included in the 2015 ERS/ATS classification criteria for IPAF (NSIP, NSIP with OP overlap, and OP) and showed the opposite results. Ito et al. [11] reported that IPAF with an NSIP pattern had higher HR than NSIP with OP/OP pattern (HR, 4.48; 95% CI, 1.28 to 15.77). However, Dai et al. [14] reported that the OP pattern had a higher HR than NSIP or NSIP with an OP pattern (HR, 3.385; 95% CI, 1.017 to 11.261). With regard to specific autoantibodies, patients with IPAF who had SSc-related autoantibodies (anti-topoisomerase antibody, ANA with a nucleolar or centromere pattern, anti-ribonucleoprotein antibody) had poorer prognoses [11,14], suggesting that a specific IPAF subgroup has a distinctive prognosis compared to other forms of IPAF. Taken together, some prognostic factors (age, baseline pulmonary function, and history of smoking) in IPAF were similar to those in IIP, and other factors (morphological patterns and specific autoantibodies) were solely associated with IPAF. Although the 2015 ERS/ATS classification criteria for IPAF were established to define IPAF and conduct studies on IPAF, physicians should consider the individual characteristics of patients with IPAF for prediction of prognosis.
CONCLUSIONS
There have been a number of IPAF-associated studies since the establishment of the classification criteria for IPAF in 2015. A specific morphological pattern is associated with prognosis, and immunosuppressants are widely used for IPAF. However, most studies were conducted retrospectively. Little is known about IPAF. Considering its complexity and heterogeneity, more well-designed clinical interventional and prospective observational studies would enhance our knowledge of IPAF. Furthermore, the 2015 classification criteria should be revised to facilitate more precise clinical research. In the diagnosis and treatment of IPAF, a multidisciplinary team approach by experts in rheumatology, pulmonology, radiology, pathology, and laboratory medicine is necessary.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Acknowledgments
This research was supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology, Republic of Korea (NRF-2018R1D1A1A02050982).