Granulocyte colony-stimulating factor in bronchoalveolar lavage fluid is a potential biomarker for prognostic prediction of idiopathic pulmonary fibrosis
Article information

Abstract
Background/Aims
Neutrophilia is frequently observed in bronchoalveolar lavage fluid (BALF) of idiopathic pulmonary fibrosis (IPF) patients. Granulocyte colony-stimulating factor (G-CSF) is a potent neutrophil-activating glycoprotein. However, the clinical implications of G-CSF remain poorly understood.in patients with IPF. Therefore, we evaluated the relationship between the G-CSF concentration in BALF and the progression of fibrosis, including in terms of the decline in lung function and long-term survival rate.
Methods
G-CSF concentrations were measured in BALF using enzyme-linked immunosorbent assay (ELISA). The survival rate was estimated using Kaplan-Meier survival analyses.
Results
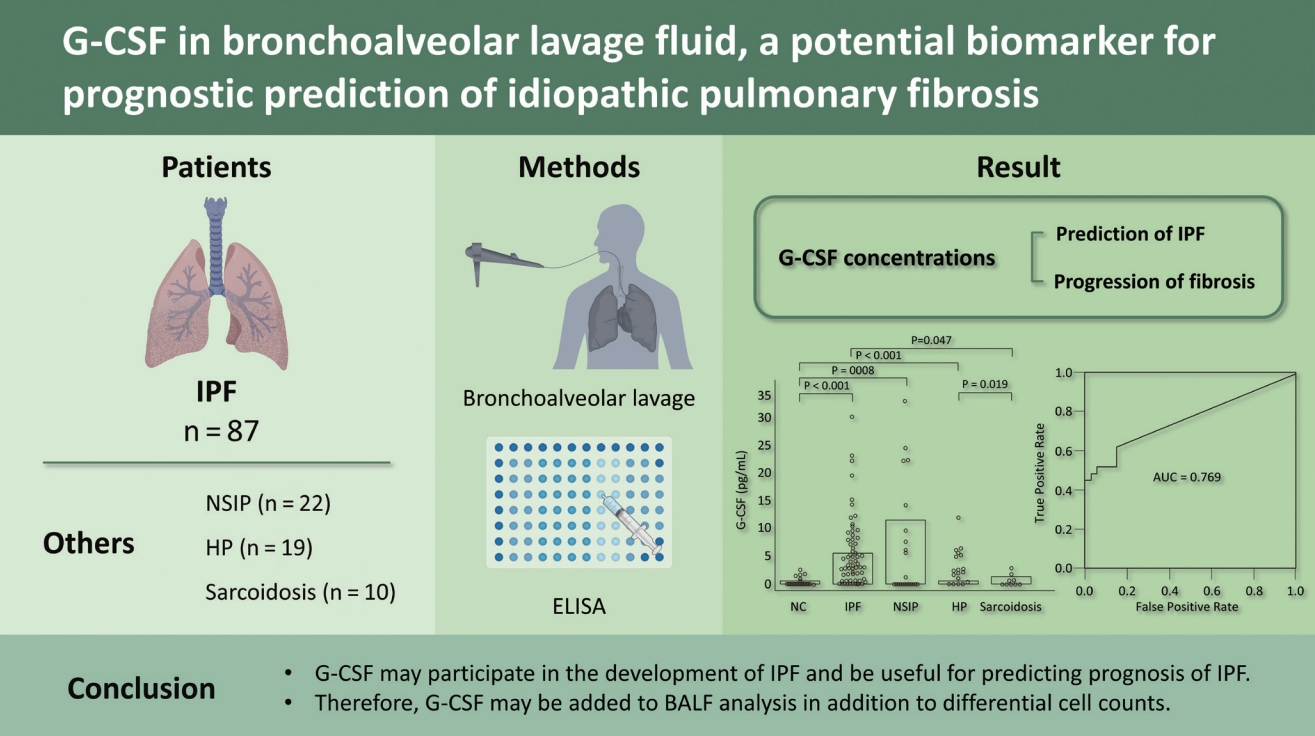
G-CSF protein levels were significantly higher in IPF (n = 87; 1.88 [0 to 5.68 pg/mL]), nonspecific interstitial pneumonia (n = 22; 0.58 [0 to 11.64 pg/mL]), and hypersensitivity pneumonitis (n = 19; 2.48 [0.46 to 5.71 pg/mL]) patients than in normal controls (n = 33; 0 [0 to 0.68 pg/mL]) (all p < 0.01). A receiver operating characteristic curve showed a difference in G-CSF levels between IPF and NC (area under the curve, 0.769): The G-CSF cut-off of 0.96 pg/mL indicated 84.9% specificity and 63.2% sensitivity for IPF. The survival rate was significantly lower in the group with G-CSF > 2.872 pg/mL than in the group with ≤ 2.872 pg/mL (hazard ratio, 2.69; p = 0.041). The annual decline in diffusing capacity of the lung for carbon monoxide was positively correlated with the G-CSF level (p = 0.018).
Conclusions
G-CSF may participate in the development of IPF and be useful for predicting the prognosis of IPF. Therefore, G-CSF should be analyzed in BALF, in addition to differential cell counts.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive form of interstitial lung disease (ILD) of unknown etiology characterized by progressive fibrosis and worsening lung function [1]. In genetically susceptible people, exposure to environmental risk factors causes repetitive micro-injury to the lung tissue and vasculature, which produce excessive amounts of pro-fibrotic mediators over anti-fibrotic mediators to trigger epithelial-mesenchymal signals leading to aberrant repair and fibrosis [2]. Fibrogenic cytokines and chemokines, such as transforming growth factor-β1, platelet-derived growth factor (PDGF), vascular endothelial growth factor, and fibroblast growth factor, have essential roles in these processes. However, immune and inflammatory cells encourage fibroblast proliferation [3]. Recruited macrophages and neutrophils produce PDGF, C-C motif chemokine ligand (CXCL) 2, macrophage-colony stimulating factor, and colony-stimulating factor 1, all of which have direct profibrotic effects [4,5]. In addition, neutrophil granules release neutrophil elastase (NE) and matrix-degrading proteins, such as matrix metalloproteinases (MMPs) [6,7]. Neutrophils also generate extracellular neutrophil traps [8], which result in the activation of fibroblasts and accumulation of the extracellular matrix (ECM) [3,9]. Among the inflammatory cells in IPF bronchoalveolar lavage fluid (BALF), the number of neutrophils predicts long-term mortality [10–12], and the levels of NE and MMPs increase markedly in BALF [13,14]. In addition, the levels of cytokeratin 19, an alveolar epithelial marker, correlate with the number of neutrophils in BALF, suggesting a relationship between neutrophilic inflammation and epithelial injury in this context [15].
The recruitment and activation of neutrophils in the lung require specific chemokines and cytokines. In humans, seven chemokines (CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8) and two chemokine receptors (CXCR1 and CXCR2) mediate neutrophil functions [16]. Additionally, granulocyte colony-stimulating factor (G-CSF) is a major extracellular regulator of hematopoiesis and the innate immune system that affects the survival, proliferation, and differentiation of all neutrophil lineage cells from hematopoietic stem cells to mature neutrophils [17]. Furthermore, G-CSF enhances the functions of monocytes, macrophages, and endothelial cells, which carry G-CSF receptors [18]. Interestingly, G-CSF differentiates murine splenic fibrocyte-like cells into monocytes and dendritic cells [19]. G-CSF concentrations are significantly elevated in BALF from patients with IPF compared to those with non-IPF lung diseases [20], suggesting that G-CSF may have specific roles in the recruitment and activation of neutrophils, monocytes, macrophages, and endothelial cells in the lungs of IPF patients [21,22].
Human G-CSF is produced not only by monocytes and macrophages but also by endothelial and bone marrow stromal cells [23]. Furthermore, fibroblasts and epithelial cells produce G-CSF when stimulated by interferon-β [24] and interleukin-17 [25]. Although in vivo administration of G-CSF has few side effects, some studies have indicated that G-CSF aggravates the lung toxicity triggered by pneumotoxic agents [26]. In contrast, G-CSF has anti-fibrotic effects on bleomycin-induced lung fibrosis through the migration of bone marrow stem cells to damaged lung tissues due to increased CXCR4 expression [27,28]. Thus, G-CSF might have dual effects on the progression of IPF. Therefore, we measured G-CSF concentrations in BALF samples to evaluate the relationship between G-CSF and the progression of fibrosis, such as the decline in lung function and the long-term survival rate, in a relatively large number of patients with IPF.
METHODS
Study subjects
BALF from patients with ILDs, including IPF, nonspecific interstitial pneumonia (NSIP), hypersensitivity pneumonitis (HP), and sarcoidosis were obtained from the Biobank of Soonchunhyang University Hospital, Bucheon, Korea after approval of the study protocol by the hospital Ethics Committee (SCHCA-IRB-2018–10–034 and 201910-BR-058). BAL was obtained within the first 2 weeks of the initial examination. All patients underwent bronchoscopy in a stable state, and none had any signs of infection.
Informed consent for study participation and sample donation was obtained from each subject. All subjects were examined by physicians and underwent a chest X-ray, high-resolution chest computed tomography (HRCT), and pulmonary function tests. The diagnosis of IPF was based on 2011 [29] and 2018 guidelines proposed by the American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Society (ATS/ERS/ JRS/ALAT) Committee on Idiopathic Pulmonary Fibrosis [30]. No subject had evidence of any underlying collagen vascular disease according to the laboratory results and symptoms. The diagnostic criteria for NSIP, HP, and sarcoidosis were based on international consensus statements [31–33]. To exclude other diseases with histological profiles similar to HP and sarcoidosis, biopsy tissues were subjected to acid-fast bacilli and Gomori methenamine silver staining to verify the absence of microorganisms and fungi. Forced vital capacity (FVC) and the diffusing capacity of the lung for carbon monoxide (DLCO) were measured every 3 months in subjects with IPF, and the annual rate of decline was estimated as [last FVC (or DLCO) – baseline FVC (or DLCO)] / baseline FVC (or DLCO) / follow-up years. The normal controls (NC) had no respiratory symptoms, as determined by a screening questionnaire [34], and had forced expiratory volume in 1 second (FEV1) and FVC > 80% predicted and normal chest radiograms.
G-CSF enzyme-linked immunosorbent assay in BALF
Bronchoalveolar lavage was performed in lung segments with the greatest disease involvement according to HRCT in patients who were not undergoing immunosuppressive therapy, or in the right middle lobe of the controls, as described previously [35]. Differential cell counts were performed on 500 BALF cells placed on slides prepared using a cytocentrifuge and Diff-Quik stain. The total cell count was measured using a hemocytometer. Cells were removed from the supernatant by centrifugation (500 g, 5 minutes), and were stored at −80°C. The G-CSF protein level was measured using an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer’s recommendations. The lower limit of detection was 2 pg/mL, and values below this limit were set to 0. The inter- and intra-assay coefficients of variation were < 15%.
Statistical analysis
Data were analyzed using SPSS version 20.0 software (IBM Co., Armonk, NY, USA). The normality of the distributions was tested using the Shapiro-Wilk test. Categorical values were compared using the chi-square test and Fisher’s exact test. Continuous values were compared using the Kruskal-Wallis test and post hoc analysis; the Mann-Whitney U test was used to compare two groups. Correlations between the G-CSF levels and other parameters were analyzed using Spearman’s correlation coefficient. Receiver operator characteristic (ROC) analysis was performed to define the cut-off values. The best cut-off value was calculated using the highest Youden index. Differences between areas under the curve (AUCs) were compared to Z tests according to De Long et al. [36] using MedCalc Statistical Software version 12.2.1.0 (MedCalc Software, Ostend, Belgium). Cox proportional hazards regression models with backward elimination were used to identify independent risk factors for IPF mortality. Survival rates were compared using Kaplan-Meier survival analyses and log-rank tests. The data are presented as the median with interquartile range for variables with skewed distributions or as the mean ± standard error of the mean for variables with normal distributions. A p < 0.05 was considered significant.
RESULTS
Clinical characteristics of the study groups
BALF was obtained from patients with IPF (n = 87), NSIP (n = 22), HP (n = 19), and sarcoidosis (n = 10). The patients with IPF had significantly more macrophages, neutrophils, and eosinophils in the BALF and lower FVC and FEV1 values compared to the NC (p < 0.05) (Table 1). The NSIP, HP, and sarcoidosis groups also had significantly (p < 0.05) higher total cell counts and numbers of macrophages, neutrophils, and eosinophils in the BALF, and lower FVC and DLCO values than the NC (Table 1).
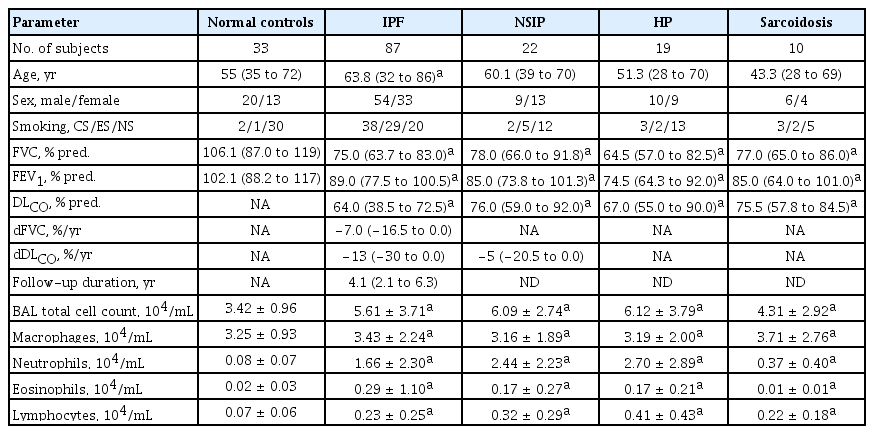
Clinical characteristics of the subjects who underwent bronchoalveolar lavage
Comparison of G-CSF levels in the BALF between IPF, other ILDs, and the NC
G-CSF protein in BALF was detected in five of 33 NCs (15%), 55 of 87 IPF patients (63%), 12 of 22 NSIP patients (54%), 15 of 19 HP patients (79%), and five of 10 sarcoidosis patients (50%). The G-CSF levels were significantly higher in the IPF (1.88 [0 to 5.68 pg/mL]), NSIP (0.58 [0 to 11.64 pg/mL]), and HP (2.48 [0.46 to 5.71 pg/mL]) groups than the NC group (0 [0 to 0.68 pg/mL]) (p < 0.01) (Fig. 1A). The G-CSF levels in the IPF and HP groups were higher than the sarcoidosis group (0.32 [0 to 1.46 pg/mL]) (p < 0.05). The ROC curve showed a clear difference between the NC and IPF groups (AUC = 0.769) (Fig. 1B). A cut-off value of 0.96 pg/mL indicated 74.1% accuracy, 84.9% specificity, and 62.3% sensitivity between the two groups.
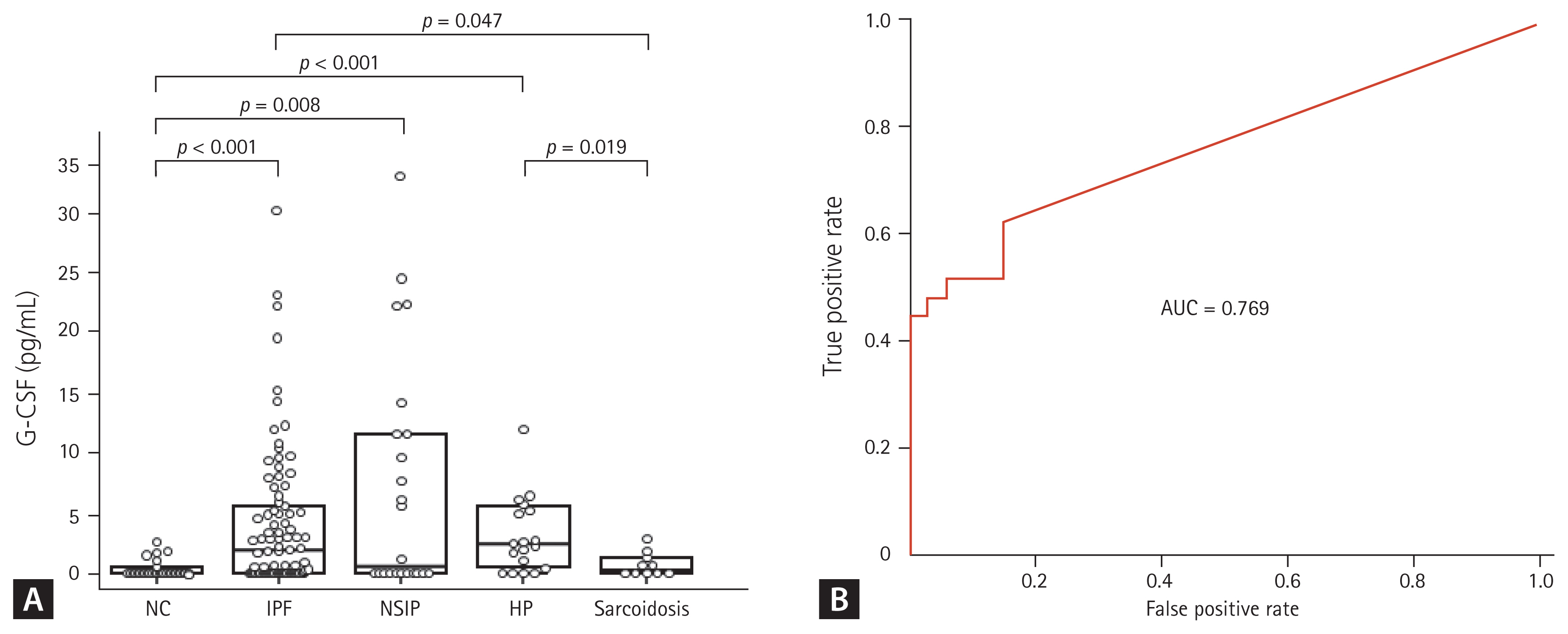
Granulocyte colony-stimulating factor (G-CSF) protein concentrations in bronchoalveolar lavage fluid and the receiver operator characteristic (ROC) curves. (A) The G-CSF protein was detected in 25 of 33 normal controls (NC), 82 of 87 idiopathic pulmonary fibrosis (IPF) patients, 20 of 22 nonspecific interstitial pneumonia (NSIP) patients, 18 of 19 hypersensitivity pneumonitis (HP) patients, and all 10 sarcoidosis patients. The data are presented as median values with interquartile range. (B) The ROC curve of the G-CSF protein concentration between the IPF and NC groups. A cut-off value of 0.96 pg/mL had 74.1% accuracy, 84.9% specificity, and 62.3% sensitivity for differentiating between the two groups. AUC, area under the curve.
Relationship between the G-CSF level and survival rate, cell number in the BALF, and clinical parameters in IPF
The mortality data of the IPF patients up to the last follow-up were collected from the hospital, and the data of 84 IPF patients who were followed for > 1 year were subjected to survival analysis. Seventeen of those patients died in our hospital, while information on survival was not available for the patients seen at other hospital; those patients were assumed to be alive at the time of the last follow-up. The 84 IPF patients followed for > 1 year were subjected to further analysis. A univariate Cox proportional hazards model showed that a high G-CSF level in the BAL fluid and low FVC values were significantly associated with an increased risk of mortality (Table 2). Multivariate analysis revealed that a high G-CSF level (hazard ratio [HR], 1.0527; 95% confidence interval [CI], 1.0027 to 1.1051; p = 0.0394) and low FVC value (HR, 0.9510; 95% CI, 0.9208 to 0.9822; p = 0.0024) were independently associated with a poor prognosis (Table 2). The Youden index was calculated based on the sensitivity and specificity of each diagnostic cut-off point on the ROC curve [36], and the point with the highest Youden index (sensitivity of 52.94% and specificity of 68.66%) was selected as the optimal G-CSF cut-off point (2.872 pg/mL) for survival. Kaplan-Meier analysis compared the survival of patients with IPF (n = 84) followed up over 10 years with low (> 2.872 pg/mL) and high (≤ 2.872 pg/mL) G-CSF levels. The survival rate was significantly lower in the group with G-CSF > 2.872 pg/mL (n = 36) than in the group with ≤ 2.872 pg/mL (n = 48; HR, 2.69; 95% CI, 1.01 to 7.15; p = 0.041) (Fig. 2). Among the clinical parameters, the neutrophil count in BALF differed significantly between the higher and lower groups using the 2.872 pg/mL G-CSF level (p = 0.001) (Table 3). G-CSF levels were positively correlated with neutrophils (r = 0.325, p = 0.002) and inversely with macrophages (r = −0.284, p = 0.008) and lymphocytes (r = −0.246, p = 0.022) (Fig. 3). Among the lung function parameters, the annual decline in DLCO (%/year) was positively correlated with the G-CSF level (r = 0.266, p = 0.018) (Fig. 3 and Supplementary Table 1), but there were no correlation between FVC or the annual decline in FVC and the G-CSF level (Supplementary Table 1).
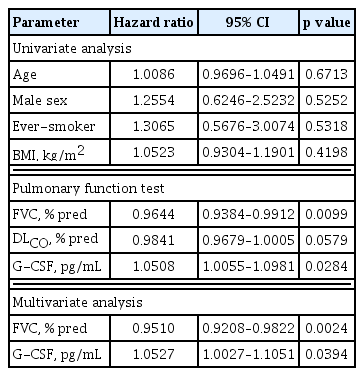
Risk factors for the mortality in patients with idiopathic pulmonary fibrosis assessed by Cox proportional hazards model
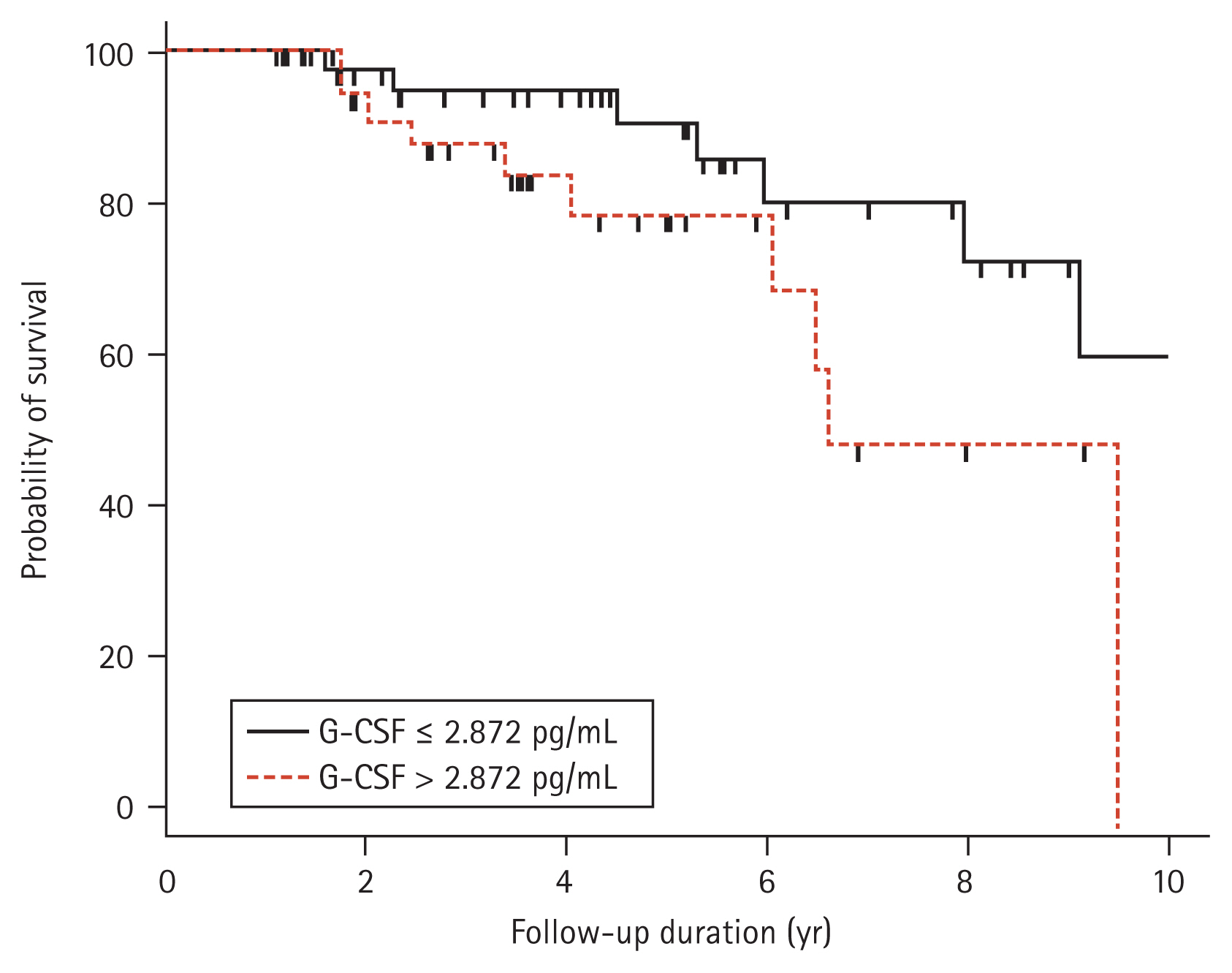
Kaplan-Meier plot of 84 subjects with idiopathic pulmonary fibrosis followed for 1 to 10 years. The percent survival was significantly lower in the group with granulocyte colony-stimulating factor (G-CSF) > 2.872 pg/mL (n = 36, dotted line) than the group with G-CSF ≤ 2.872 pg/mL (n = 48, solid line) (hazard ratio, 2.69; 95% confidence interval, 1.01 to 7.15; p = 0.041).
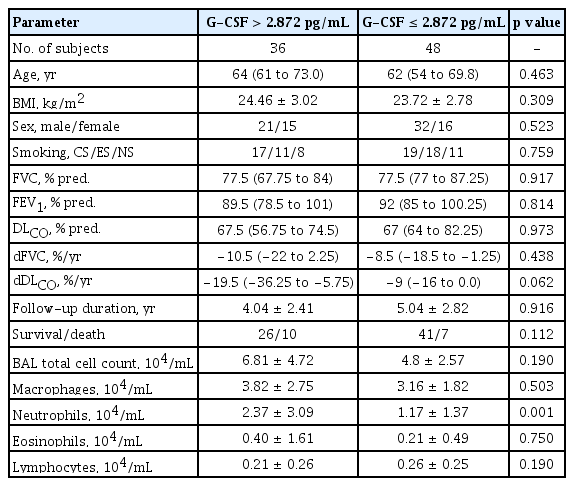
Clinical characteristics of patients with idiopathic pulmonary fibrosis classified according to the levels of G-CSF and percentage of neutrophils in bronchoalveolar lavage fluid
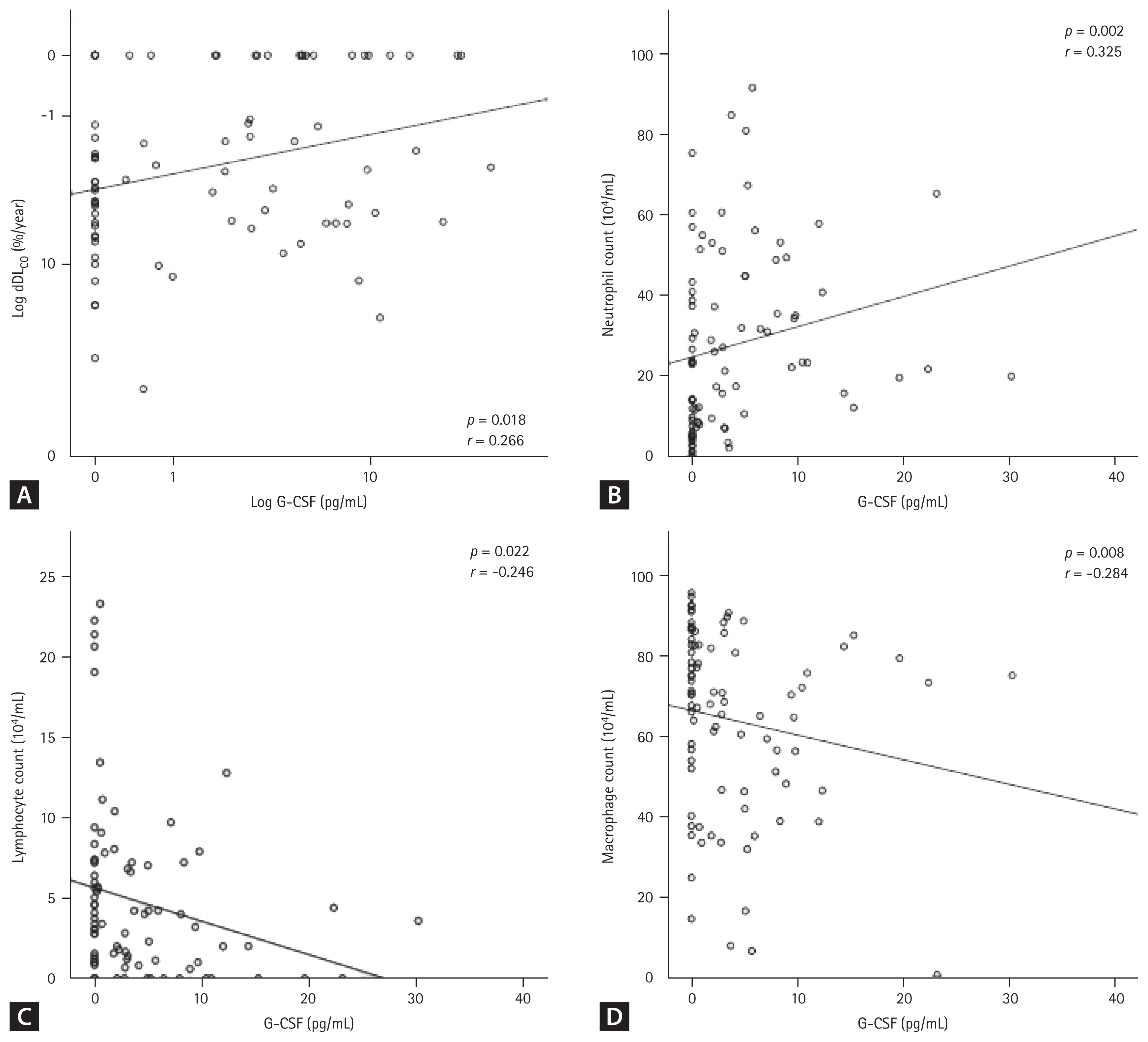
Correlation between granulocyte colony-stimulating factor (G-CSF) levels and cell number in bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis. G-CSF protein concentrations were positively correlated with (A) diffusing capacity of the lung for carbon monoxide (DLCO; %/year) (p = 0.018, r = 0.226) and (B) neutrophil number (p = 0.002, r = 0.325) but negatively correlated with (C) lymphocytes (p = 0.022, r = −0.246) and (D) macrophages (p = 0.008, r = −0.284).
DISCUSSION
We verified high G-CSF levels in IPF BALF compared to NC and a cut-off value of 0.96 pg/mL with 84.9% specificity and 62.3% sensitivity for discriminating the two groups. Ashitani et al. [20] revealed high G-CSF levels in 24 patients with IPF compared to 16 NC. They also reported that G-CSF was high in patients with other lung diseases, including Wegener’s granulomatosis, bacterial pneumonia, asthma, bronchiectasis, and acute respiratory distress syndrome (ARDS). In our study, the G-CSF level increased in the BALF of patients with NSIP and HP. The increase in the G-CSF level was correlated with the number of neutrophils in BALF on both studies, indicating that a high G-CSF level in the lungs is not specific to IPF or other ILD, but occurs in other diseases with neutrophilic inflammation. Notably, the G-CSF concentration predicted the survival rate of the patients with IPF. Furthermore, the G-CSF levels were positively correlated with the decrease in DLCO. Accordingly, G-CSF levels may be a surrogate biomarker predicting the prognosis of IPF.
The mechanism behind the relationship between G-CSF and the prognosis of IPF may be as follows. G-CSF overproduction leads to lung neutrophilia, which releases NE and MMPs [6]. NE degrades the ECM into collagens I–IV, fibronectin, laminin, and elastin. It also induces the proliferation of fibroblasts and differentiation of myofibroblasts [37,38]. To estimate the effects of neutrophilia in the lungs, the survival rate was analyzed according to the level of BAL neutrophils: subjects with > 19.4% neutrophils had a significantly lower survival rate than those with ≤ 19.4% (p = 0.024) (Supplementary Fig. 1A). Thus, the survival rate based on G-CSF was reanalyzed after adjusting for the neutrophil count, and the difference in survival rate remained significant (p = 0.041) (Supplementary Fig. 1B). This indicates that G-CSF on its own may have harmful effects during the progression of IPF.
Although G-CSF pulmonary toxicity is very rare in healthy volunteers, pulmonary G-CSF toxicity is relatively common in combination with other pneumotoxic agents. A meta-analysis of 21 studies reported 73 cases of G-CSF-related pulmonary toxicity in neutropenic patients who received cyclophosphamide, bleomycin, or methotrexate [26]. Of these, 36 patients had IP with mild to moderate hypoxemia and 35 had ARDS. This result indicates that a high G-CSF level can accelerate the process of pulmonary fibrosis. G-CSF may also enhance the functions of monocytes, macrophages, and endothelial cells, which carry G-CSF receptors [18]. By binding to these receptors, G-CSF regulates the production of cytokines, such as tumor necrosis factor-α, produced by macrophages, which further potentiates lung damage [37]. Thus, G-CSF appears to lower the dose threshold for pulmonary toxicity of chemotherapy drugs, and this may be cumulative and more common after multiple courses of chemotherapy. In the present study, G-CSF levels and survival were reanalyzed after the use of cytotoxic drugs (Supplementary Fig. 2): 26 subjects used azathioprine, while 58 subjects did not, and the survival rates were not different between the groups. Many clinical, radiological, and physiological predictors are associated with survival in patients with IPF [1]. There are also several models, including the Gender-Age-Physiology (GAP) Index that predict mortality in patients with IPF [38]. The GAP Index stratifies patients into three stages based on clinical (sex and age) and physiological (FVC and DLCO) variables. Our results showed that the G-CSF protein levels and the GAP Index were not related in patients with IPF (Supplementary Fig. 3). These data suggest that the G-CSF concentration is an independent risk factor regardless of clinical or physiological factors.
Our study had several limitations. First, we did not measure other neutrophilic active cytokines or chemokines in the BALF from the study subjects. The neutrophil chemotactic activity in BALF from patients with IPF is reduced by 35% after neutralization with anti-human G-CSF antiserum [20], indicating that other factors besides G-CSF could trigger an inflammatory response by recruiting and activating neutrophils [21,22]. Second, there were too few patients with other ILDs, including NSIP, HP, and sarcoidosis to evaluate the diagnostic significance. Finally, validation studies using other cohorts are required to improve the diagnostic utility of G-CSF.
In conclusion, considering the poor prognosis of IPF and the recent development of inhibitors of disease progression (pirfenidone [39] and nintedanib [40]), clinicians must make early and accurate predictions of the long-term prognosis. Our study revealed the utility of G-CSF as a predictive biomarker for prognosing IPF. Therefore, G-CSF could be added to the BALF analysis in addition to the differential cell count.
KEY MESSAGE
1. The granulocyte colony-stimulating factor (G-CSF) level increased in the bronchoalveolar lavage fluid (BALF) of patients with idiopathic pulmonary fibrosis (IPF).
2. The survival rate decreased in IPF patients with G-CSF > 2.872 pg/mL.
3. The G-CSF level in BALF may be useful for prognostic prediction of IPF.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2020R1I1A1A01067088), and a Soonchunhyang University grant to Jong Sook Park. The biospecimens and data used for this study were provided by the Biobank of Soonchunhyang University Bucheon Hospital, a member of the Korea Biobank Network (KBN4_A06).