Long-acting interferon: pioneering disease modification of myeloproliferative neoplasms
Article information

Abstract
Myeloproliferative neoplasms (MPNs) are clonal disorders of hematopoietic stem cells. The malignant clones produce cytokines that drive self-perpetuating inflammatory responses and tend to transform into more aggressive clones, leading to disease progression. The progression of MPNs follows a biological sequence from the early phases of malignancy, polycythemia vera, and essential thrombocythemia, to advanced myelofibrosis and leukemic transformation. To date, the treatment of MPNs has focused on preventing thrombosis by decreasing blood cell counts and relieving disease-related symptoms. However, interferon (IFN) has been used to treat MPNs because of its ability to attack cancer cells directly and modulate the immune system. IFN also has the potential to modulate diseases by inhibiting JAK2 mutations, and recent studies have demonstrated clinical and molecular improvements. Long-acting IFN is administered less frequently and has fewer adverse effects than conventional IFN. The current state of research on long-acting IFN in patients with MPNs is discussed, along with future directions.
INTRODUCTION
Myeloproliferative neoplasms (MPNs) are a cluster of disorders marked by abnormal growth and proliferation of bone marrow myeloid progenitor cells [1]. In MPNs, stem cells produce abnormally high numbers of blood cell lineages. If the Philadelphia chromosome exists, the MPN is classified as chronic myeloid leukemia (CML). Otherwise, it is referred to as Philadelphia chromosome-negative MPN, which includes polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). MPNs are inflammatory neoplasms in which a malignant clone generates cytokines that self-perpetuate the inflammatory drive [2]. Their progression follows a biological sequence from the early phases of malignancy (ET/PV) to advanced MF and leukemic transformation. Mutations in JAK2, CALR, and MPL play significant roles in the development of MPNs, with additional mutations contributing to disease progression [3]. Symptoms of MPN, such as fatigue, headache, general weakness, and itchiness, are caused by pro-inflammatory cytokines, which contribute to the development of thrombosis and hemorrhage, as well as myeloid cell proliferation [4]. The treatment goals for patients with ET and PV are managing the symptoms, preventing vascular complications, and monitoring for progression to leukemia [5]. Antiplatelet agents such as low-dose acetylsalicylic acid and cytoreductive agents such as hydroxyurea (HU) and anagrelide are the most common treatment options in Korea. In patients with PMF, JAK inhibitors such as ruxolitinib and fedratinib ameliorate symptoms, decrease splenomegaly, and correlate with an increased survival compared with conventional therapy. Currently, allogeneic hematopoietic stem cell transplantation is the only potentially curative treatment for PMF [6]. In general, patients with ET/PV have a less severe disease burden than those with MF. The incidence of thrombosis, MF/leukemic transformation, and mortality remain low in patients with PV and ET. However, patients with ET/PV outnumber those with PMF, and their disease progression is of considerable interest.
Interferon (IFN) was identified as a cytokine that inhibits viral replication approximately 65 years ago [7]. Since then, its antitumor effects and ability to modulate immunity have been recognized and it is used to treat solid and hematologic malignancies. In 1986, the Food and Drug Administration (FDA) approved IFN alpha (IFNα) for the treatment of hairy cell leukemia as the first immunotherapeutic drug [8]. IFNα was once the front-line treatment standard for CML until tyrosine kinase inhibitors targeting the BCR/ABL fusion protein were developed [9]. In patients with Philadelphia chromosome-negative MPNs, IFNα modulates symptoms via antiproliferative, proapoptotic, and immunomodulatory effects, and decreases vascular events. However, frequent doses and adverse events are significant obstacles to its widespread long-term use. The introduction of more tolerable forms of IFNα, such as pegylated IFNα (peg-IFNα) and, more recently, ropeginterferon alfa-2b (ropeg-IFNα-2b), has increased the interest in therapies [10]. IFNα maintains the JAK2 molecule in a relatively reduced state, which has disease-modifying potential and actual clinical outcomes have recently been reported [11,12]. This review discusses studies of long-acting IFNα and its disease-modifying potential.
MODE OF ACTION OF IFN
IFNs are classified as type I, II, or III based on the receptor and downstream signaling pathways. Type I IFN signals via JAK1 and TYK2 kinases and activates the STAT1, STAT2, and IRF9 transcription factors, leading to increased stimulation of the interferon-stimulated response element. Type I IFNs include multiple subtypes of IFN-α and a single type of IFN-β, in addition to the less well-characterized IFN-δ, -ɛ, -κ, -τ, -ω, and -ζ subtypes [13]. IFN regulates the expression and secretion of various cytokines, which in turn regulate the cellular and enzymatic pathways involved in cell proliferation, differentiation, and function [14]. IFN induces proapoptotic effects by inducing tumor necrosis factor-related apoptosis-inducing ligand and/or Fas/FasL to interact with their respective receptors. IFN can also stimulate the activity of host immune cells, such as T lymphocytes, monocytes, NK cells, and macrophages [13]. Although numerous studies have examined the pleiotropic effects of IFN, including its antiproliferative, anti-apoptotic, and immunomodulatory properties, the mechanism of action of IFN is still not completely understood. Moreover, the reasons why some patients respond to IFN while others do not remain unknown. However, Saleiro et al. [15] recently identified a pathway involving the PKC-dependent phosphorylation of ULK1 at serine residues 341 and 495, which is required for the subsequent activation of p38 MAPK. They demonstrated that this pathway is necessary for IFN-suppressive effects on primary malignant erythroid precursors from patients with MPN, and that elevated ULK1 and p38 MAPK levels correlate with the clinical response to IFN therapy in these patients [15]. Nevertheless, further investigations of the mechanisms and effects of IFN are necessary.
IFNα IN PHILADELPHIA CHROMOSOME-NEGATIVE MPNS
An initial study described the use of IFNα, which has antiproliferative effects on hematopoietic precursors, particularly the megakaryocytic lineage, for controlling thrombocytosis in patients with MPNs [16,17]. Subsequent studies confirmed that IFNα can inhibit myeloproliferation in Philadelphia chromosome-negative MPNs, reduce the need for phlebotomies, alleviate bothersome symptoms, normalize elevated leukocyte and platelet counts, and reduce spleen size [2,18]. Additionally, improvement in the degree of bone marrow fibrosis has been reported in several patients [19]. After the JAK2V617F mutation was identified in 2005, IFNα has been reported to induce molecular remission in patients with JAK2-positive MPN [20,21]. Long-term IFNα treatment maintained the molecular response in some patients [22]. These findings indicate that IFNα therapy has the potential to modify disease in some patients with MPNs. Despite these advantages, conventional IFNα is not used extensively due to its many adverse effects and requirement for frequent administration. Long-acting IFNα (peg-IFNα and ropeg-IFNα-2b) with fewer adverse effects and increased administration intervals has been introduced. Table 1 lists recent large studies of long-acting IFN [19,22–26]. Peg-IFNα has been used to treat hepatitis; however, due to the recent advent of direct-acting antiviral agents, the use of peg-IFNα as a treatment for hepatitis has decreased [27]. Peg-IFNα is currently unavailable in Korea due to supply issues. Ropeg-IFNα-2b is a mono-pegylated IFN with an extended half-life, allowing administration every other week. The FDA has approved ropeg-IFNα-2b for use as a treatment in patients with PV, making it the first IFN drug licensed for treating MPNs by the FDA. A clinical trial (SURPASS-ET, NCT04285086) comparing ropeg-IFNα-2b with anagrelide as second-line therapy for high-risk ET is ongoing [28].
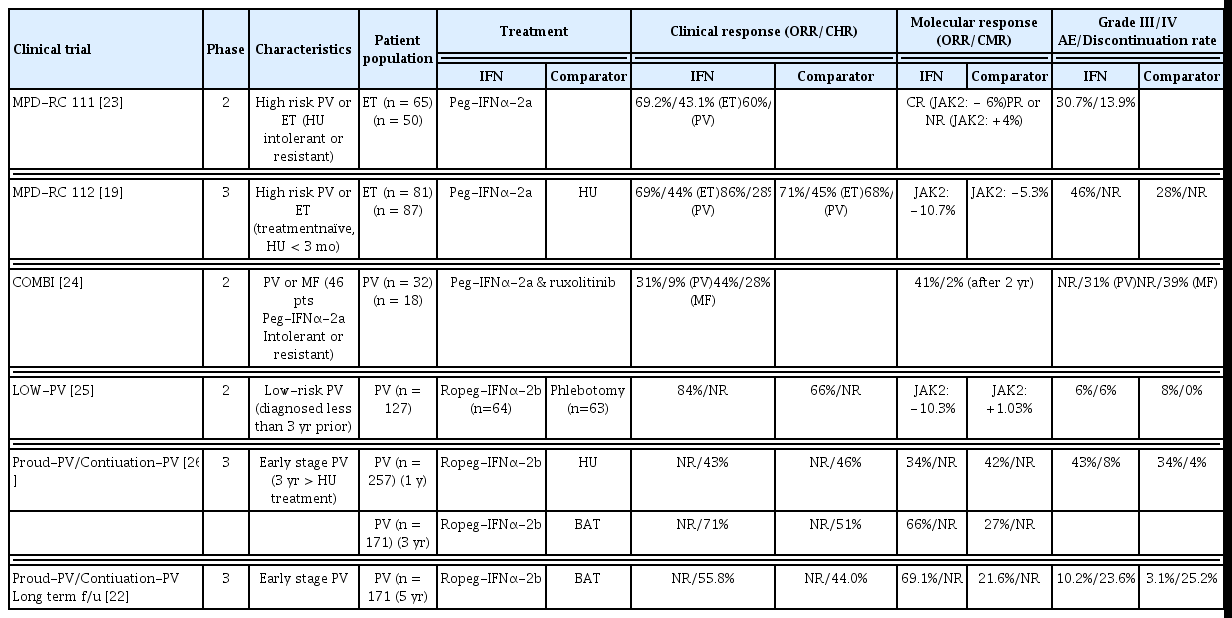
Summary of clinical trials of long-acting IFNα in patients with MPN
It is difficult to conclude that the hematologic response of long-acting IFNα is significantly superior to that of HU, especially during the early period of long-acting IFN therapy when the dosage is gradually increased to the appropriate level [26]. Furthermore, adverse events are still a concern, albeit reduced in long-acting IFNα compared to conventional IFNα. However, IFNα is excellent at achieving the optimal molecular response to reduce JAK2 variant allele frequency (VAF). Specifically, HU lost its molecular response after 1 year, whereas long-acting IFNα maintained the response after prolonged use [19,26]. In the Proud-PV/Continuation-PV 5-year follow-up, the molecular response rate was significantly higher in the ropeg-IFNα-2b arm than in the control arm (69.1 vs. 21.6%, respectively; p < 0.0001) [22]. Of importance, the median JAK2V617F VAF declined continuously during treatment with ropeg-IFNα-2b and a significant reduction in the median JAK2V617F VAF was observed in low-risk PV patients on a fixed 100 μg dose [25].
CLINICAL SIGNIFICANCE OF THE JAK2 MUTATION
The JAK2V617F point mutation was identified in Philadelphia chromosome-negative MPNs in 2005 [29]. In this point mutation, phenylalanine replaces valine in codon 617 of the JAK2 gene. It has been identified in patients diagnosed with PV, affecting 90% or more of PV patients, as well as being present in nearly 50% of patients diagnosed with ET and PMF. Hematopoietic stem cells harboring this mutation produce excess cytokines and reactive oxygen species, which modify the bone marrow niche, reinforce the growth advantage of clonal cells, and promote genomic instability and MF. The JAK2V617F mutation also affects peripheral blood cell counts [30]. When evaluating JAK2V617F VAF in PV as a continuous variable, the white blood cell (WBC) count and hematocrit were positively correlated with VAF, whereas the mean corpuscular volume and platelet count were negatively correlated with VAF [31]. Numerous clinical trials have shown that the molecular response is enhanced when the hematological response is good. Further, JAK2V617F VAF is associated with a risk of thrombosis, progression to fibrosis, and overall survival (OS) [30]. Guglielmelli et al. [32] reported that a JAK2V617F VAF > 50% was independently associated with a higher risk of venous thromboembolism in patients with both low- and high-risk PV stratified by age and thrombosis history. Lee et al. [33] reported that a high JAK2V617F VAF (≥ 58%) was associated with a poor OS in patients with PV. Several of these studies were retrospective and based on JAK2V617F VAF at the time of diagnosis or at specific time points. However, JAK2V617F VAF can change continuously throughout disease progression and treatment. Recent prospective clinical trials have reported changes in JAK2V617F VAF [22]. Further studies need to determine the clinical significance of these changes.
DISEASE MODIFYING POTENTIAL OF IFNα IN PATIENTS WITH MPNS
In patients with ET and PV, the incidence of progression to MF and leukemia is minimal, warranting extended periods of observation. Therefore, demonstrating a significant benefit in reducing the risk of disease progression in studies related to MPNs is exceedingly challenging. Therefore, IFNα should alter the course of the disease by consistently reducing JAK2V617F VAF; however, no study has yet demonstrated clinical benefit. Recent studies have demonstrated the disease-modifying potential of IFNα. A single-center retrospective analysis of 470 patients with PV compared the myelofibrosis-free survival (MFS) and OS in patients treated with IFNα with HU or phlebotomy-only (PHL-O). The patients were classified based on the initial cytoreductive therapy received for at least 1 year. In 262 low-risk patients with PV, the 20-year MFS for IFNα, HU, and PHL-O was 84, 65, and 55%, respectively (p < 0.001). In 208 high-risk patients with PV, the 20-year OS for IFNα, HU, and PHL-O was 66, 40, and 14%, respectively (p = 0.016). In the multivariate analysis, a prolonged duration of IFNα treatment was associated with a decreased incidence of MF (hazard ratio [HR], 0.91; p < 0.001) and lower mortality (HR, 0.94; p = 0.012) [12]. Although this was a retrospective study and did not reflect treatment changes after the first year, it is valuable as it reports improvements in OS and MFS over a follow-up period exceeding 10 years. In addition, the Proud-PV/Continuation-PV 6-year follow-up results announced at the European Hematology Association 2022 Congress are intriguing, although they have not been formally published [11]. The JAK2 reduction was significantly greater in the IFN group than in the controls. The molecular response in patients treated with ropeg IFNα-2b was still significantly better, compared to the controls. Event-free survival (risk events: disease progression, death, and thromboembolic events) was significantly higher in patients treated with ropeg IFNα-2b than in the controls (risk events reported in 5/95 vs. 12/74 patients, respectively; p = 0.04). This was the first prospective clinical trial showing that ropeg-IFNα-2 therapy improves the clinical outcome in patients with MPNs.
IFNα FOR PATIENTS WHO ARE RESISTANT TO OR INTOLERANT OF HU
Early laboratory studies of HU showed that it caused congenital malformations in pregnant experimental animals [34,35]. Therefore, HU should be avoided in pregnant patients, although blood cell count control is necessary for these patients who are at high risk of miscarriage due to microthrombosis from uncontrolled blood cell counts. IFNs are relatively safe for pregnant patients and fetuses, so switching from HU to IFN agents is recommended if cytoreductive agents are needed in pregnant patients [36].
HU is generally well-tolerated, even in patients who receive it for prolonged periods. However, some patients experience serious side effects, such as drug fever, pneumonia, mucosal ulcers, and skin ulcers, which may require drug discontinuation [37]. In addition, some patients develop “HU resistance”, in which the blood cell counts are not adequately controlled by HU. These HU resistant/intolerant patients are thought to have a poor survival rate compared to other patients, due to the increased risk of vascular complications and evolution into more advanced disease [38,39]. Therefore, resistant and intolerant patients should consider second-line drugs.
Although it is possible to regulate blood cell counts with available second-line drugs, further research is needed to determine whether they can prevent the evolution into advanced disease [40,41]. According to molecular genetic studies of resistant patients, resistance occurs as additional genetic mutations accumulate [42]. These additional mutations are related to the secondary malignant progression of the disease [43,44]. For second-line drugs to inhibit the evolution into more advanced disease, they must be able to overcome these additional mutations. From this perspective, long-acting IFN has the potential to reduce the burden of mutant genes, so it is an attractive choice for these patients.
EFFECTIVE ADMINISTRATION STRATEGIES FOR LONG-ACTING IFNα
Ropeg-IFNα-2b is the only available long-acting IFN for patients with MPN in Korea. This is a novel mono-pegylated IFN with pharmacokinetic properties that permit administration every 2–4 weeks. In the Proud-PV/Continuation-PV study, ropeg-IFNα-2b was started at a dose of 100 μg (50 μg for patients receiving HU) and increased by 50 μg every 2 weeks to a maximum of 500 μg until a hematologic response was observed [26]. The mean effective dose of ropeg-IFNα-2b was attained after approximately 16.2 weeks. In the Proud-PV/Continuation-PV study, ropeg-IFNα-2b was not superior to HU in the hematologic and molecular responses at the 12-month assessment. However, after 36 months, its performance improved notably. Gradual dosage escalation was implemented due to the insufficiently promising outcomes observed following a 12-month course of ropeg-IFNα-2b therapy. Recent ongoing investigations have used a 250–350–500 μg ropeg-IFNα-2b dosing titration regimen every 2 weeks. Overall, these dosing schedules are effective, and the side effects are tolerable [45]. After 3 years of the Proud-PV/Continuation-PV study, half of the patients were able to switch from a 2-week to a 3–4-week dose schedule. In other words, should the side effects remain tolerable, promptly escalating the ropeg-IFNα-2b dose can yield optimal therapeutic outcomes. Furthermore, if this favorable effect endures, prolonging the intervals between doses might enhance the tolerability of ropeg-IFNα-2b therapies. While few studies on the discontinuation of IFNα have been published, some reports have indicated that more profound molecular responses are linked to extended periods of disease remission subsequent to the discontinuation of IFNα. [46,47].
FUTURE DIRECTION OF LONG-ACTING IFNα IN PATIENTS WITH MPN
Elderly patients diagnosed with MPNs generally have a less favorable prognosis than their younger counterparts [48]. However, when comparing the mortality rates of healthy individuals and patients with MPN in the same age group, the relative risk in the younger group was greater than in the older group [49]. Thus, the treatment of young patients necessitates considerable care [50,51]. Long-acting IFNα, which is effective at regulating blood cell counts and alleviating symptoms, has been shown to have the potential to modify the disease by decreasing JAK2V617F VAF and is anticipated to play a significant role. In addition, IFNα can be administered relatively early in the progression of MPN, because many additional mutations have been identified in the group that is resistant to IFN treatment [52]. A thorough investigation is required to discover the optimal dosing schedule for more effective use by improving the tolerability of long-acting IFNα, and to maintain remission after discontinuation.
Currently, ropeg-IFNα-2b is not covered by national health insurance in Korea, but insurance coverage is required for pregnant women with MPNs who cannot use HU and for PV patients who are intolerant/resistant to HU because there are no other suitable treatments for those patients.
CONCLUSION
Patients with ET or PV have an extended life expectancy using cytoreductive therapy; however, there is an unmet need for medications that improve the disease course. Long-acting IFNα is an attractive option for patients who are intolerant or resistant to HU because IFNα has the potential to modify the disease course, reduce the risk of vascular complications and disease evolution, and improve survival. Patients who are intolerant of or resistant to HU have few treatment options, and IFNα could offer an effective new way to manage their disease. In the future, the indications for long-acting IFNα are likely to expand, and long-acting IFNα could be used effectively in many patients with MPNs.
Notes
CRedit authorship contributions
Seug Yun Yoon: conceptualization, methodology, formal analysis, writing - original draft; Sung-Yong Kim: conceptualization, data curation, writing - review & editing
Conflicts of interest
The author discloses no conflicts.
Funding
None