


INTRODUCTION
Henoch-Sch├Čnlein Purpura(HSP) is one of the leukocytoclastic vasculitis affecting small vessels and manifesting as palpable purpura and is frequently associated with systemic manifestations, such as fever, arthralgia, abdominal pain, edema, hematuria and proteinuria1,2).
Causative factors were not precisely described. We describe a case of HSP associated with disseminated tuberculosis.
CASE REPORT
A 41-year-old man was admitted to our hospital because of generalized purpura and productive cough. He was well until 2 months earlier when he developed productive cough with whitish mucoid sputum and exertional dyspnea. At that time, he was admitted to a local hospital and treated with antituberculous agents for pulmonary tuberculosis confirmed by positive sputum for acid-fast bacilli (AFB). Forty five days before entry, he noticed generalized purpura, more prominent in the lower extremities, and mild pretibial edema. Ten days before admission, high fever and generalized weakness occurred in spite of antituberculosis medication.
He was a government employee and nonsmoker. His past medical history was noncontributory. There was no history of previous nausea, vomiting, abdominal pain, constipation, diarrhea, weight loss and arthralgia.
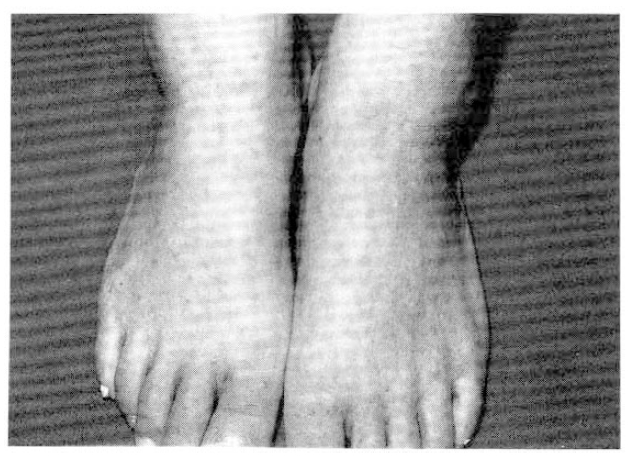
On examination, he was febrile state, temperature 38.1 ┬░C, with a blood pressure of 130/90mmHg, a respiratory rate of 20 per minute, and a heart rate of 112 beats per minute. Multiple, 0.5 ├Ś 0.5cm sized, nontender cervical lymph nodes were detected. The conjunctivae were pale. Coarse breathing sounds were heard over the both lung with rale. His bowel sounds were normal and there was no evidence of hepatosplenomegaly. Numerous nontender palpable purpura, located primarily on the lower extremities, were present with pretibial pitting edema (Fig. 1).
The chest roentgenogram taken on admission revealed diffuse scattered, ill defined nodules in entire both lung fields and patchy and confluent density in both upper lungs(Fig. 2). Abdominal ultrasonogram showed increased echoes of renal parenchyme with upper normal size. Others were not remarkable.
The hemoglobin was 9.0g/dl, with 2.8% reticulocytes. The hematocrit was 27.2%. The white blood cell count was 14.3 ├Ś 109/L, with a differential of 75% neutrophils, 20% lymphocytes, 3% monocytes, 1% eosinophil and 1% atypical lymphocyte. Wintrobe erythrocyte sedimentation rate was 65 mm/hr(corrected 18 mm/hr). The platelet count was 438 ├Ś 109/L. The antiplatelet antibody was negative. The prothrombin time and partial thromboplastin time were within normal limits. Blood chemistry revealed total serum protein 5.8 g/dl, albumin 1.7 g/dl, AST 65 U/L, ALT 23 U/L, triglyceride 109 mg/dl, BUN 22.5 mg/dl, creatinine 1.5 mg/dl, calcium 7.6 mg/dl and phosphate 5.1 mg/dl with normal serum electrolyte. The serum Ig A was 640 mg/dl, Ig G 2020 mg/dl, Ig M 150 mg/dl and Ig E 831 IU/ml. The serum C3, C4 were 162, 37.8 mg/dl, respectively. The urinalysis was reported as showing 300 mg/dl of proteinuria, 20ŌĆō25 red blood cells, 3ŌĆō5 white cells per high-power field and WBC cast. Sixty-five percent of the red cells were dysmorphic. In a 24-hour specimen of urine, protein was 6930 mg, creatinine 693 mg. A serum protein electrophoresis revealed hypoalbuminemia with increased gamma globulin fraction and urine electrophoresis showed protein losing pattern. Stool specimen gave a negative test for occult blood. The tests for antistreptolysin O titer, ANA, LE cell, VDRL, hepatitis B surface antigen and antibody and rheumatoid factor were all negative.
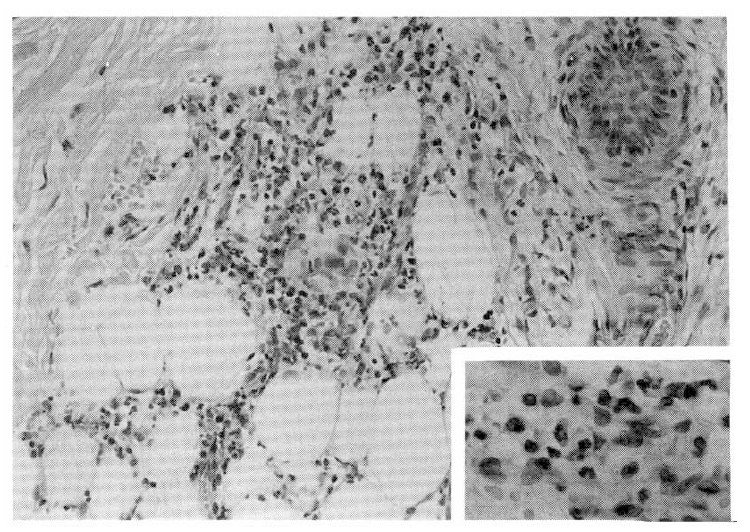
Cultures of blood remained sterile. The arterial blood gas showed pH 7.37, PCO2 32.4 mmHg. PO2 71.6 mmHg, HCO3 18.9 mmol/L. The pulmonary function studies showed that FEV1 was 2.04 L(62% predicted). Three separate smears of sputum for AFB were negative. The antituberculous chemotherapy was begun with isoniazid (300 mg), ethambutol (800 mg), rifampin (450 mg) and pyrazinamide (1.5 g), all once daily with pyridoxine. A punch biopsy specimen of the palpable purpuric lesions on the posterior aspect of the leg was consistent with the diagnosis of ŌĆ£leukocytoclastic vasculitisŌĆØ (Fig. 3). The hospital course was complicated by continued hematuria and proteinuria. The skin lesions slowly resolved by antituberculous agents. The patient was discharged nineteen days later and then readmitted five weeks later for renal biopsy.
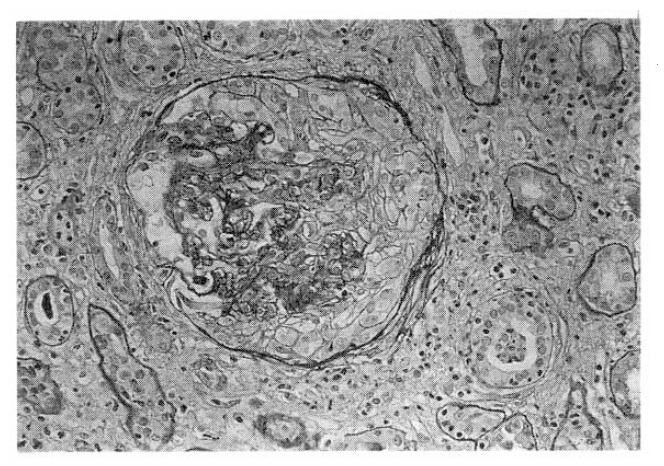
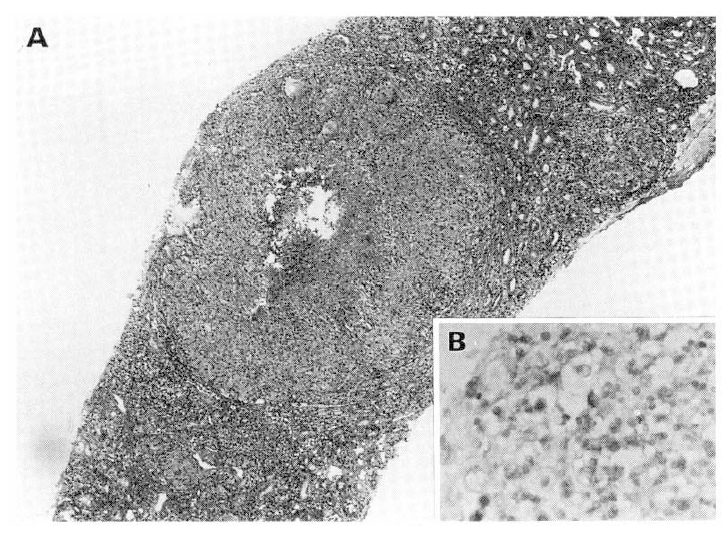
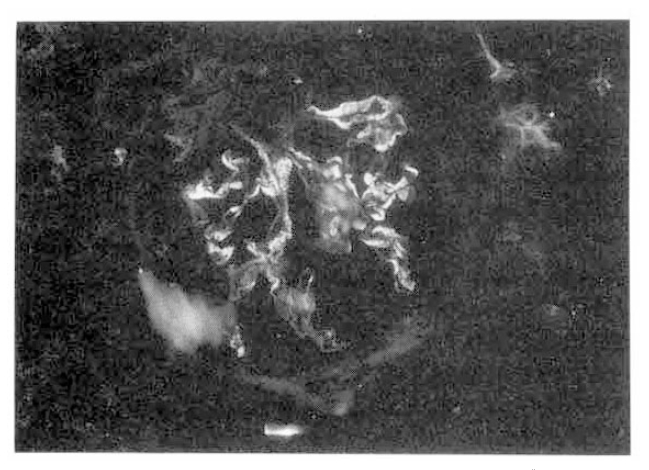
On readmission, clinical signs were improved and the pulmonary lesions on chest X-ray film were slightly cleared. Renal biopsy was performed. Light microscopy demonstrated mesangial cell hyperplasia and widening of mesangial matrix. There were fibrous crescents associated with adhesion of BowmanŌĆÖs capsule (Fig. 4). There was the evidence of interstitial chronic granulomatous inflammation with caseous necrosis, with moderate fibrosis, lymphoplasmacytic infiltration, compatible with renal involvement of tuberculosis(Fig. 5). Immunofluorescent findings were strong nodular mesangial Ig A deposit with trace granular Ig G deposition along peripheral capillary loops and perivascular C3 deposit(Fig. 6).
Thereafter, antituberculous drugs were administered for 12 months. He completely recovered from the pulmonary and renal manifestations and remained symptom free in 24 months after antituberculous drugs started.
DISCUSSION
In this patient with disseminated tuberculosis, palpable purpura were noticed on lower extremities and proteinuria accompanying hematuria occurred several days after antituberculous chemotherapy. He was compatible with systemic vasculitis, Henoch-Sch├Čnlein purpura(HSP), based on the cutaneous leukocytoclastic vasculitis and mesangial Ig A, Ig G and C3 deposits revealed by skin and renal biopsy1,2).
The precise etiology of HSP is unknown, although many causes have been proposed. Numerous causative factors or associated disorders include infectious diseases such as upper respiratory infection with beta-hemolytic Streptococcus, subacute bacterial endocarditis with Staphylococcus, B viral hepatitis and acquired immune deficiency syndrome(AIDS), immunization, hypersensitivity reactions to drug or food, cryoglobulinemia, systemic lupus erythematosus and malignant tumors2ŌĆō8).
In this case, drugs might have contributed to the development of HSP. But drugs were not a possible triggering factor because his pulmonary tuberculosis, skin lesions and renal manifestations were controlled by drugs. Renal biopsy showed chronic granulomatous interstitial inflammation with caseous necrosis and, therefore, infection itself rather than drugs might be the implicating factor for the genesis of HSP.
The vasculitis of HSP is leukocytoclastic vasculitis based on the histopathologic appearance with a clinical picture characterized by palpable purpura, typically on the lower extremities. The histologic features required to make the diagnosis include small-vessel destruction characterized by a transmural inflammatory infiltrate consisting of neutrophils and nuclear fragments, endothelial swelling, fibrin deposition, and, often, erythrocyte extravasation9ŌĆō12).
A number of clinical reviews have recently published that immune complexes may play a primary role in the pathogenesis of Henoch-Sch├Čnlein vasculitis. Initially, the mechanism of disease activity is the deposition of immunoglobulin or immune complexes in the blood vessel walls, leading to activation of alternative complement cascade and subsequent generation of chemotactic factors with infiltration of neutrophils. During the course of phagocytosis of these immune complexes, release of destructive lysosomal enzymes could then result in injury to vessel walls, thrombosis and hemorrhage. Dermal and mesangial deposits of Ig A, Ig G, Ig M, complement and fibrin were reported9,10,13,14). Stevenson et al reported that granular deposits of Ig A have been found in small-vessel walls of the glomerular mesangium. They also demonstrated the same pattern of immune deposits in each site, by immunofluorescent microscopy, consisting principally of Ig A and C3, which ultrastructurally appeared as electrondense deposits. They speculated that the selective organic injury produced by Ig A deposits in HSP may result from an excessive immune stimulation, from an abnormality of Ig A production or from inability to clear Ig A polymers. But the site of formation of the immune complexes and factors that determine where they are deposited are not clearly understood15).
Although we did not perform a test for circulating immune complexes, concurrent appearance of deposition of immunoglobulin and complement by direct immunofluorescent assay would suggest that there be evidence of circulating immune complexes in serum formed by exogenous or endogenous antigenic stimulation9).
In general, the prognosis of HSP is good, although it has a remarkable tendency to recur several times during a period of weeks or months, and treatment usually consists of supportive and symptomatic therapy until resolution. This patient has been followed-up with fully recovered purpuric skin rash and impressive improvement of pulmonary lesions on chest x-ray. Also, there was no evidence of aggravation or recurrence of any manifestation of HSP during the follow-up period. Follw-up laboratory findings, 2 months later, showed total serum protein 7.1 g/dl, albumin 3.3 g/dl and serum Ig A 377 mg/dl, Ig G 1680 mg/dl, Ig M 143 mg/dl and Ig E 10 IU/Ml. The serum C3, C4 were 146, 31.8 mg/dl, respectively. In a 24-hour specimen of urine (2000 Ml), protein was 4560 mg, creatinine 644 mg. It was only antituberculous therapy that was enough to cure HSP in our patient.
Shribman et al. reported the case of a man with miliary tuberculosis and focal proliferative glomerulonephritis(GN)16). Meyrier et al. described the association between membranoproliferative GN in the course of disseminated tuberculosis17). Rodriguez-Garcia et al. experienced a patient with pulmonary tuberculosis and membranous nephropathy18). As in earlier reported cases, a coincidence between a common disease, such as tuberculosis, and a systemic vasculitis such as HSP should be considered. We, however, believe that a causative factor of this case was disseminated tuberculosis.
In summary, we conclude that disseminated tuberculosis might be the cause of HSP with renal involvement in this case because antituberculous therapy abolished both disseminated tuberculosis and manifestations of HSP.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





